

Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease
- PMID: 15509540
- PMCID: PMC1618661
- DOI: 10.1016/S0002-9440(10)63427-X
Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease
Abstract
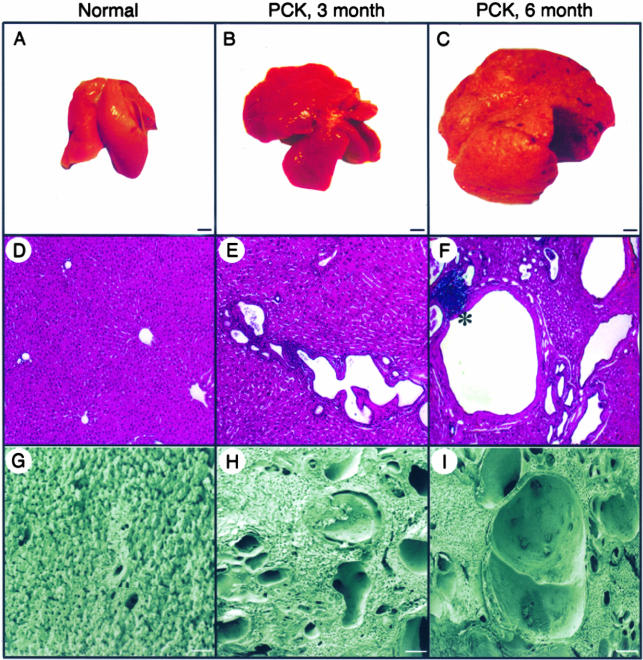
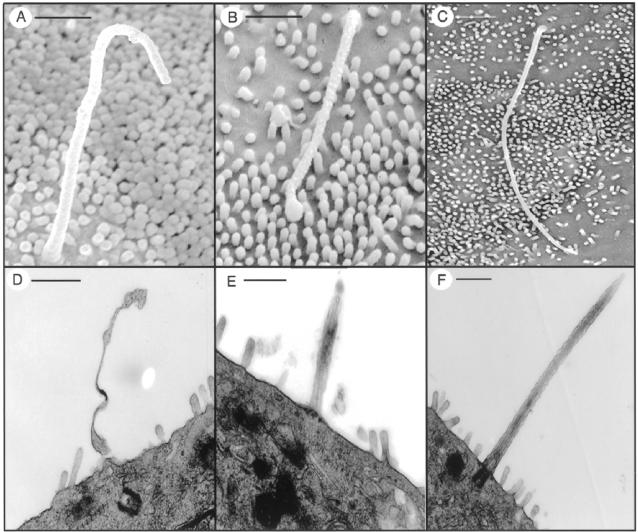
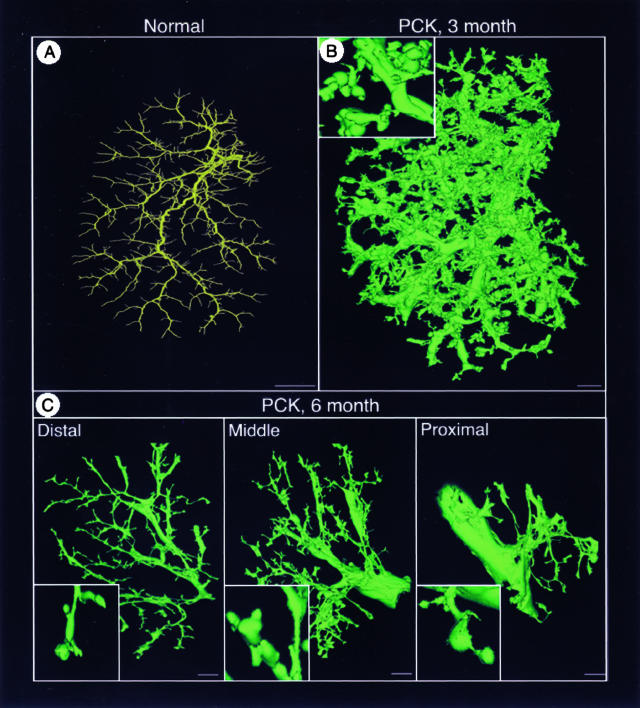
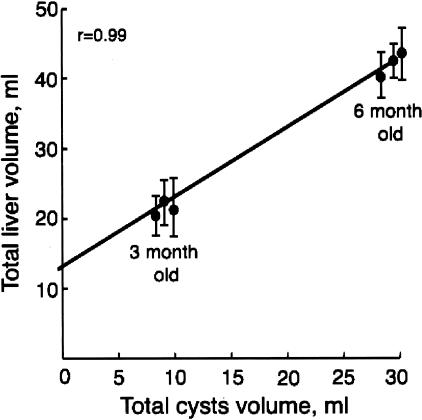
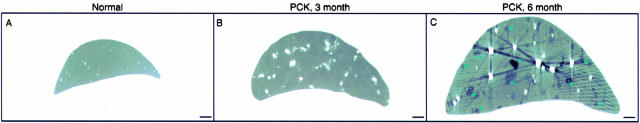
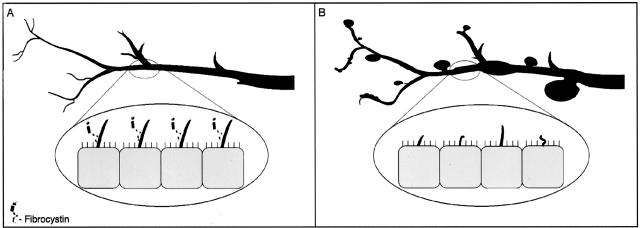
Hepatic polycystic disease occurs alone or in combination with polycystic kidney disease (PKD). In autosomal recessive PKD (ARPKD), liver lesions are the major cause of morbidity and mortality in older patients. ARPKD is caused by a mutation to PKHD1 and the PCK rat is an orthologous model of disease. Recently, we showed that fibrocystin, Pkhd1 protein, is localized to primary cilia in rat cholangiocytes and that disruption of its ciliary expression results in biliary cystogenesis. This study describes biliary phenotype in the PCK rat using micro-computed tomography scanning and three-dimensional reconstruction, and light, scanning, and transmission microscopy. Our results show that the biliary tree undergoes extensive remodeling resulting in bile duct dilatation, focal budding, and formation of cysts that are initially connected to bile ducts, but throughout time separate from them. Progressive liver enlargement results from massive cyst formation while liver parenchymal volume remains unchanged. Cilia in cystic cells are abnormal consistent with the notion that the primary defect in ARPKD resulting in cystogenesis may be linked to ciliary dysfunction. Our results suggest that the PCK rat is a useful model for studies of biliary cystogenesis and treatment options of inherited cystic liver disease.
Figures




References
-
- Iglesias DM, Palmitano J, Arrizurieta E, Kornblihtt AR, Herrera M, Bernath V, Matrin RS. Isolated polycystic liver disease not linked to polycystic kidney disease 1 and 2. Dig Dis Sci. 1999;44:385–388. - PubMed
-
- Pirson Y, Lannoy N, Peters D, Guubel A, Gigot J-F, Breuning M, Verellen-Dumoulin C. Isolated polycystic liver disease as a distinct genetic disease, unlinked to polycystic kidney disease 1 and polycystic kidney disease 2. Hepatology. 1996;23:249–252. - PubMed
-
- Drenth JPH, teMorsche RHM, Smink R, Bonifacino JS, Jansen JBMJ. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33:345–347. - PubMed
-
- Calvet JP, Grantham JJ. The genetic and physiology of polycystic kidney disease. Semin Nephrol. 2001;21:107–123. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
