

Dopamine modulates release from corticostriatal terminals
- PMID: 15509741
- PMCID: PMC6730145
- DOI: 10.1523/JNEUROSCI.2891-04.2004
Dopamine modulates release from corticostriatal terminals
Abstract
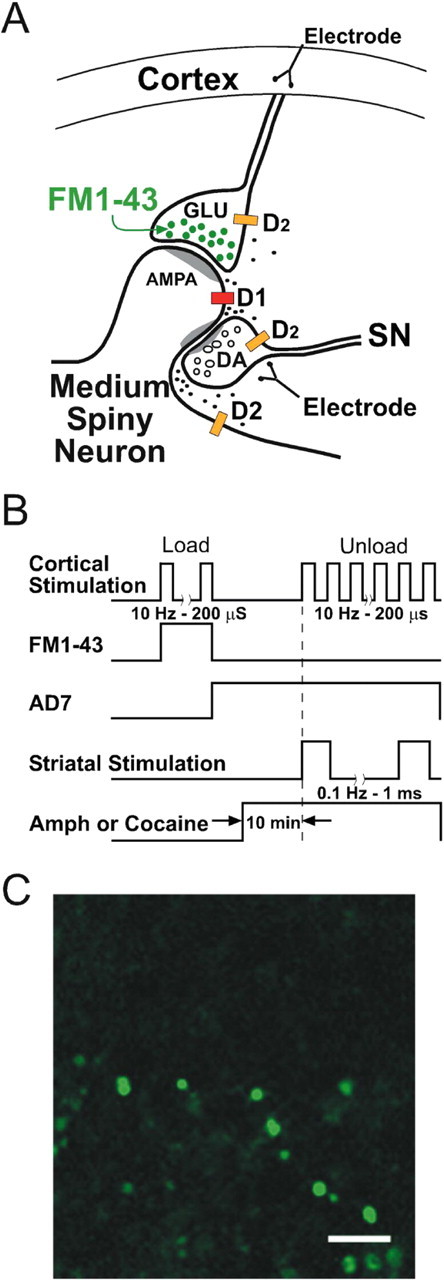
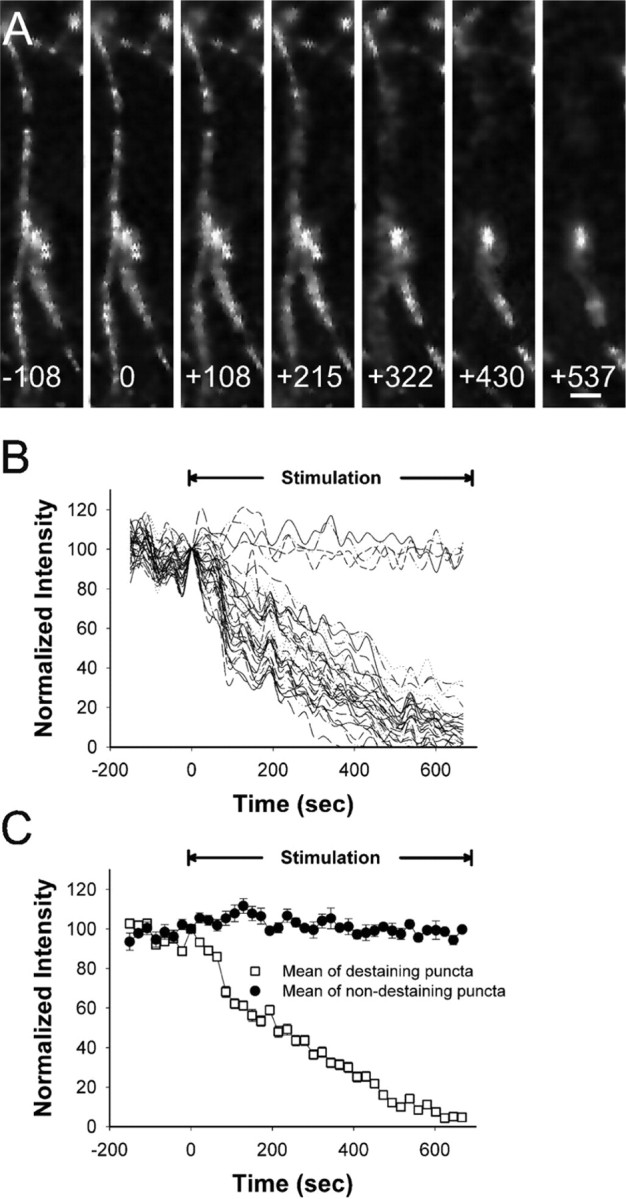
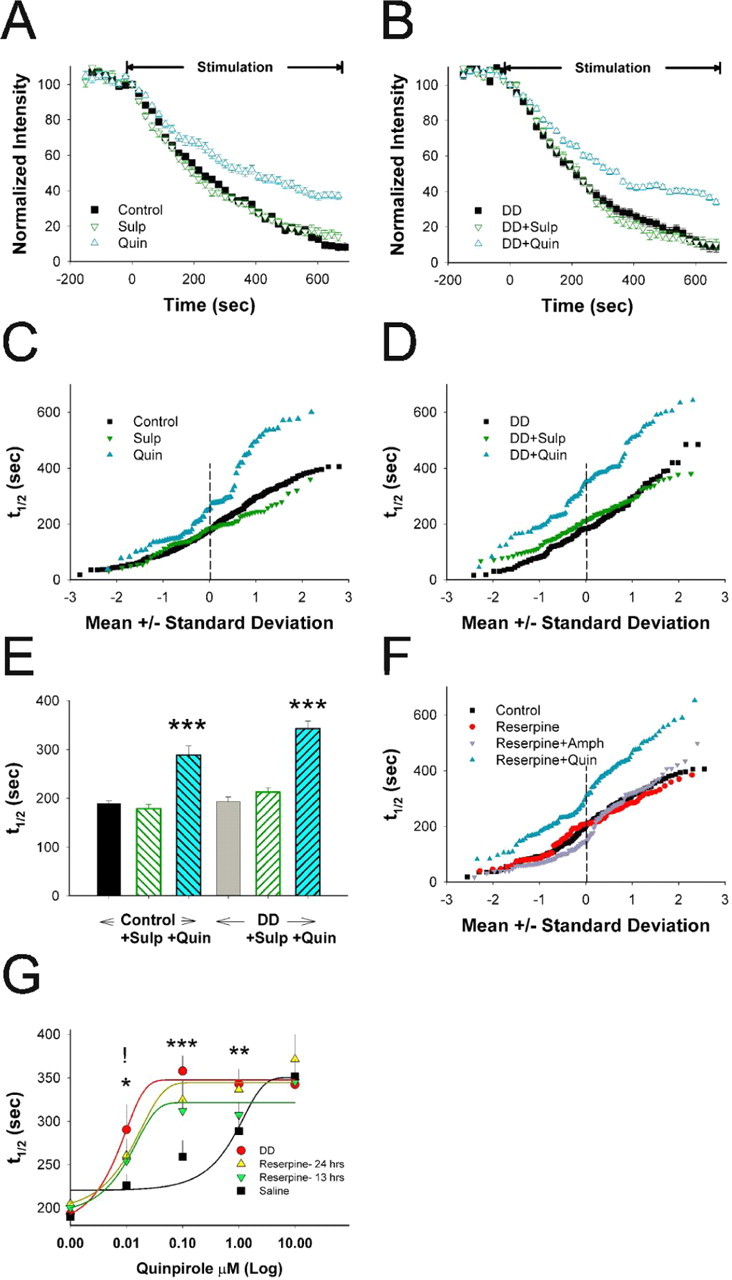
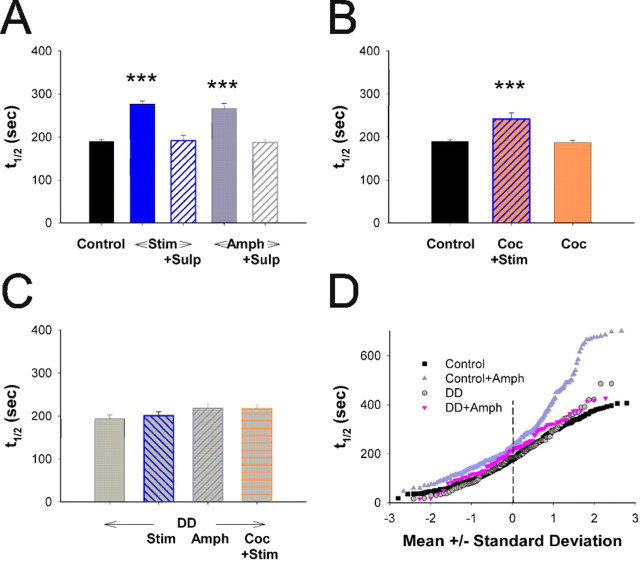
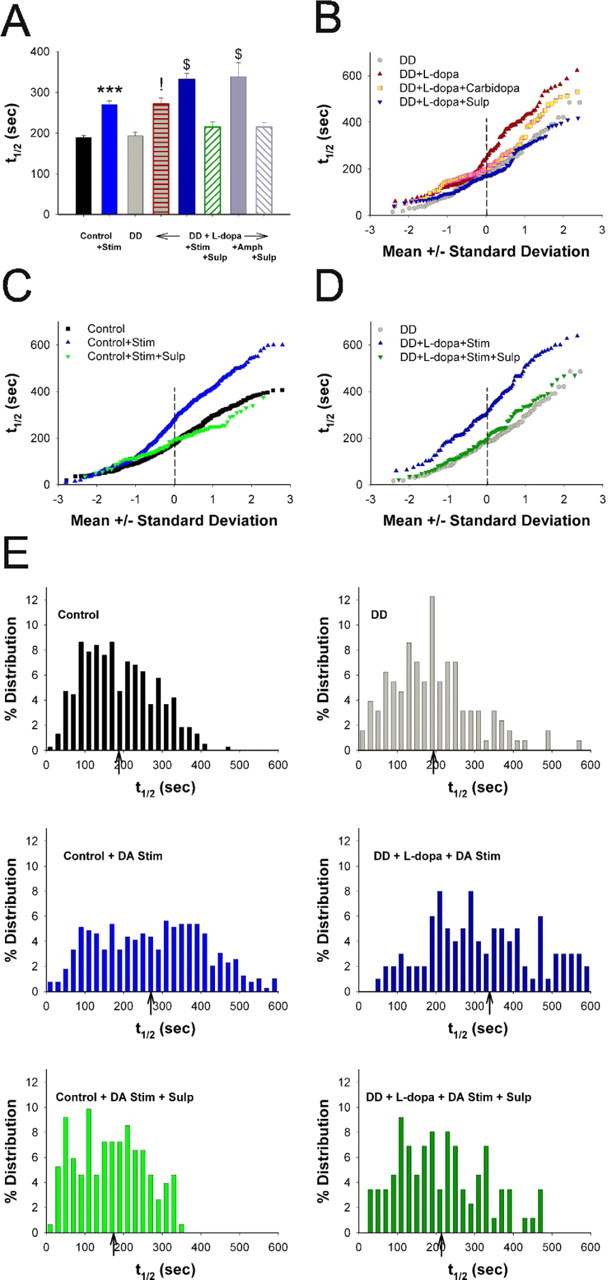
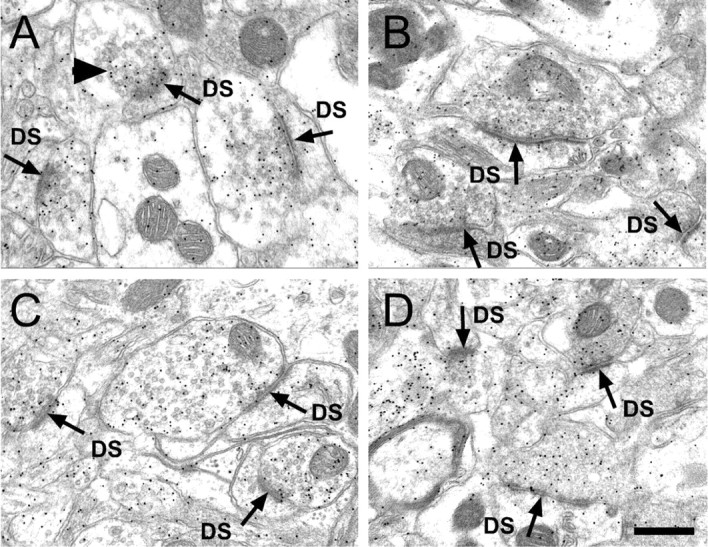
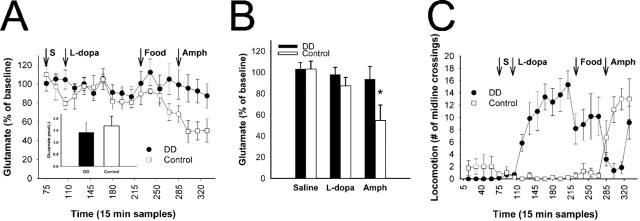
Normal striatal function is dependent on the availability of synaptic dopamine to modulate neurotransmission. Within the striatum, excitatory inputs from cortical glutamatergic neurons and modulatory inputs from midbrain dopamine neurons converge onto dendritic spines of medium spiny neurons. In addition to dopamine receptors on medium spiny neurons, D2 receptors are also present on corticostriatal terminals, where they act to dampen striatal excitation. To determine the effect of dopamine depletion on corticostriatal activity, we used the styryl dye FM1-43 in combination with multiphoton confocal microscopy in slice preparations from dopamine-deficient (DD) and reserpine-treated mice. The activity-dependent release of FM1-43 out of corticostriatal terminals allows a measure of kinetics quantified by the halftime decay of fluorescence intensity. In DD, reserpine-treated, and control mice, exposure to the D2-like receptor agonist quinpirole revealed modulation of corticostriatal kinetics with depression of FM1-43 destaining. In DD and reserpine-treated mice, quinpirole decreased destaining to a greater extent, and at a lower dose, consistent with hypersensitive corticostriatal D2 receptors. Compared with controls, slices from DD mice did not react to amphetamine or to cocaine with dopamine-releasing striatal stimulation unless the animals were pretreated with l-3,4-dihydroxyphenylalanine (l-dopa). Electron microscopy and immunogold labeling for glutamate terminals within the striatum demonstrated that the observed differences in kinetics of corticostriatal terminals in DD mice were not attributable to aberrant cytoarchitecture or glutamate density. Microdialysis revealed that basal extracellular striatal glutamate was normal in DD mice. These data indicate that dopamine deficiency results in morphologically normal corticostriatal terminals with hypersensitive D2 receptors.
Figures
References
-
- Akopian G, Walsh JP (2002) Corticostriatal paired-pulse potentiation produced by voltage-dependent activation of NMDA receptors and L-type Ca(2+) channels. J Neurophysiol 87: 157-165. - PubMed
-
- Albin RL, Young AB, Penney JB (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12: 366-375. - PubMed
-
- Arbuthnott GW, Ingham CA, Wickens JR (1998) Modulation by dopamine of rat corticostriatal input. Adv Pharmacol 42: 733-736. - PubMed
-
- Arnt J (1985) Behavioural stimulation is induced by separate dopamine D-1 and D-2 receptor sites in reserpine-pretreated but not in normal rats. Eur J Pharmacol 113: 79-88. - PubMed
-
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D (2004) Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron 42: 653-663. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources