

Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A
- PMID: 15510216
- PMCID: PMC526470
- DOI: 10.1038/sj.emboj.7600455
Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A
Abstract
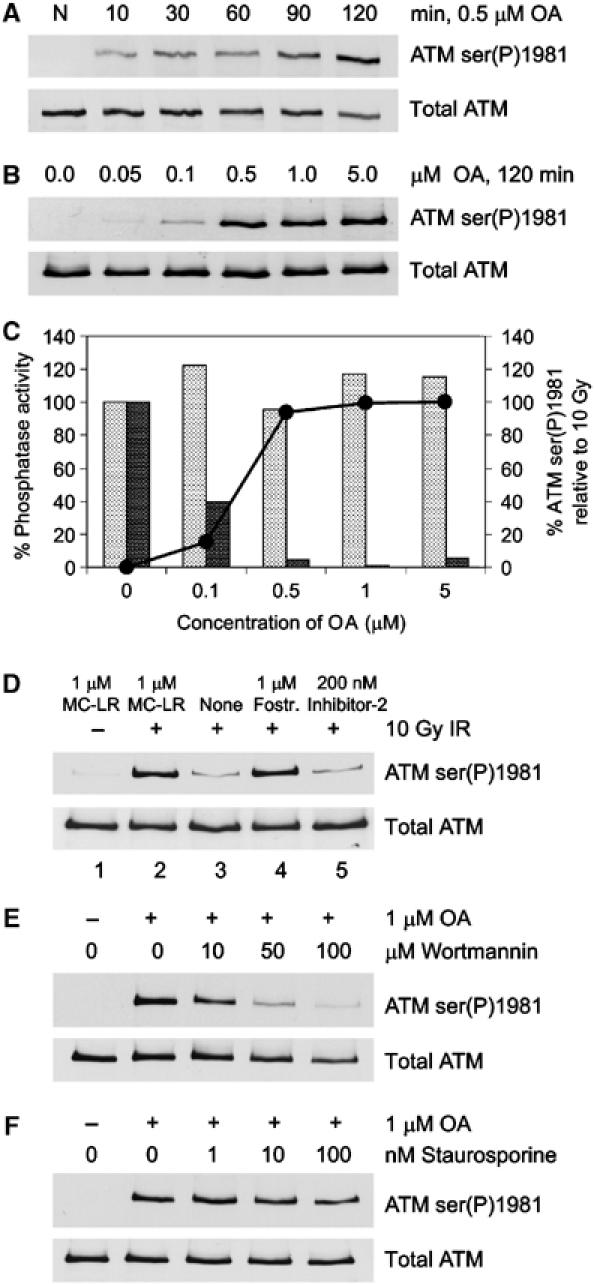
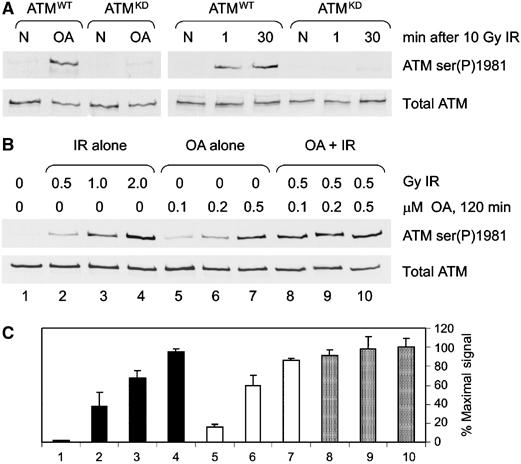
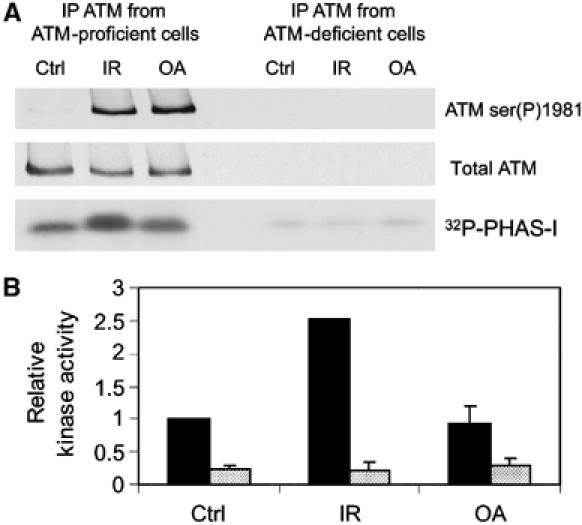
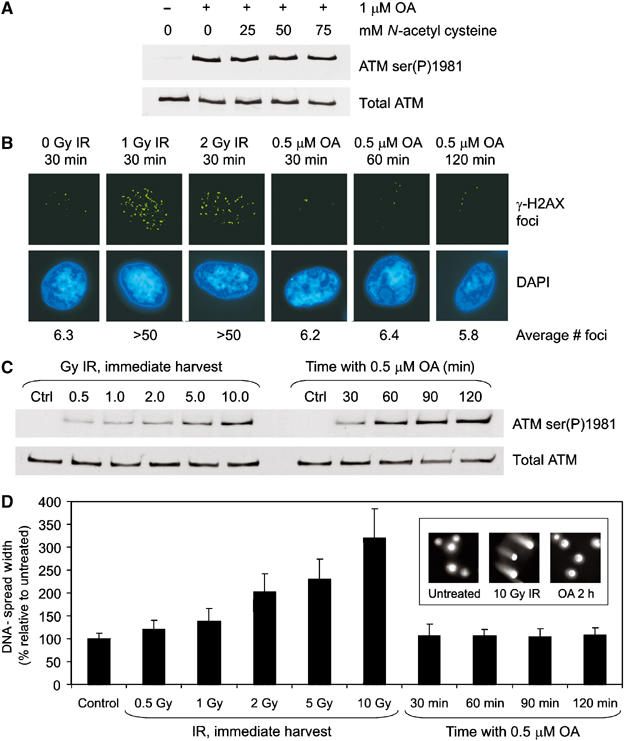
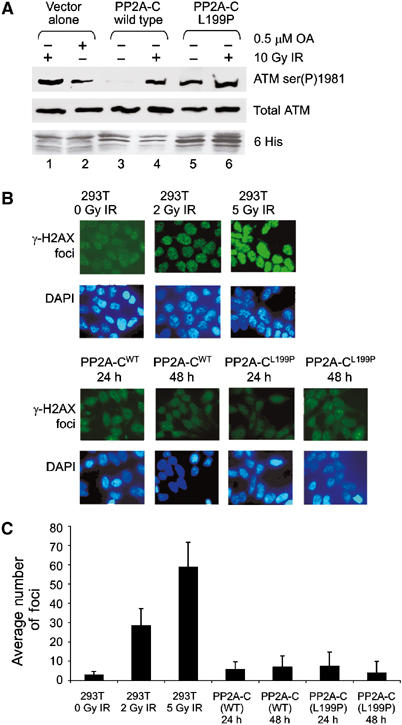
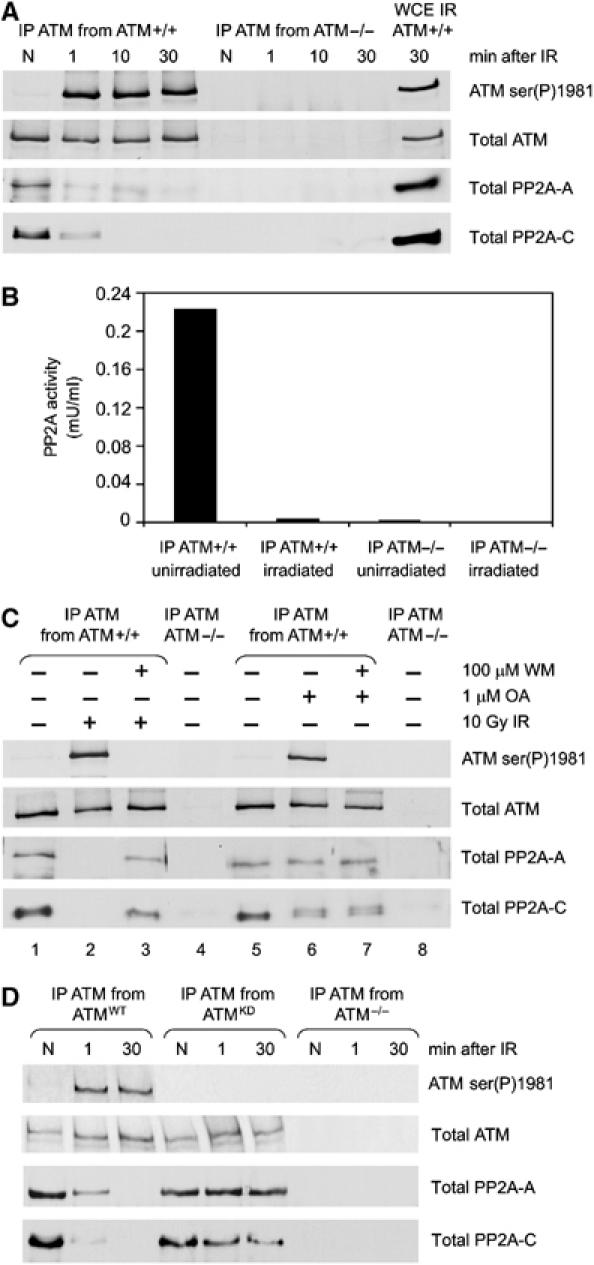
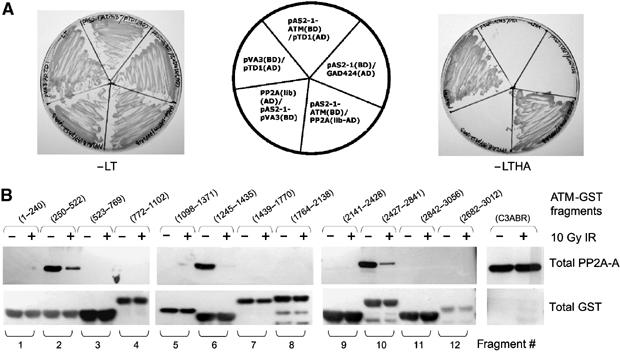
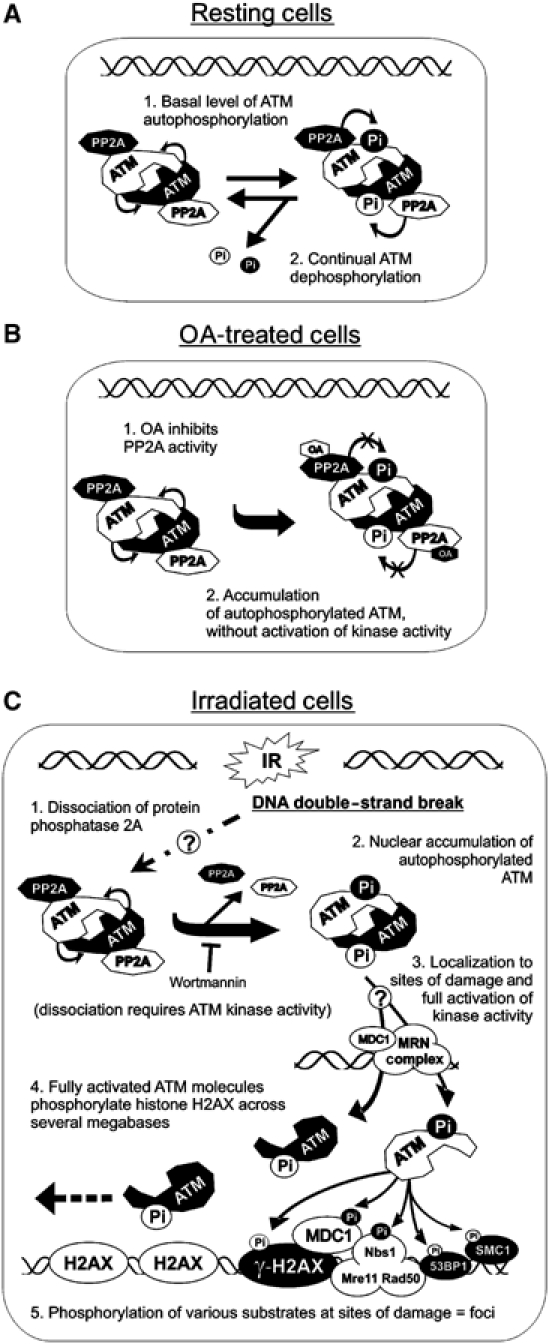
Ionizing radiation induces autophosphorylation of the ataxia-telangiectasia mutated (ATM) protein kinase on serine 1981; however, the precise mechanisms that regulate ATM activation are not fully understood. Here, we show that the protein phosphatase inhibitor okadaic acid (OA) induces autophosphorylation of ATM on serine 1981 in unirradiated cells at concentrations that inhibit protein phosphatase 2A-like activity in vitro. OA did not induce gamma-H2AX foci, suggesting that it induces ATM autophosphorylation by inactivation of a protein phosphatase rather than by inducing DNA double-strand breaks. In support of this, we show that ATM interacts with the scaffolding (A) subunit of protein phosphatase 2A (PP2A), that the scaffolding and catalytic (C) subunits of PP2A interact with ATM in undamaged cells and that immunoprecipitates of ATM from undamaged cells contain PP2A-like protein phosphatase activity. Moreover, we show that IR induces phosphorylation-dependent dissociation of PP2A from ATM and loss of the associated protein phosphatase activity. We propose that PP2A plays an important role in the regulation of ATM autophosphorylation and activity in vivo.
Figures
Similar articles
-
Utilizing protein phosphatase inhibitors to define PP2A as a regulator of ataxia-telangiectasia mutated.Methods Mol Biol. 2007;365:47-59. doi: 10.1385/1-59745-267-X:47. Methods Mol Biol. 2007. PMID: 17200553
-
Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX.Mol Cell Biol. 2010 Mar;30(6):1368-81. doi: 10.1128/MCB.00741-09. Epub 2010 Jan 11. Mol Cell Biol. 2010. PMID: 20065038 Free PMC article.
-
Protein phosphatase 2A antagonizes ATM and ATR in a Cdk2- and Cdc7-independent DNA damage checkpoint.Mol Cell Biol. 2006 Mar;26(5):1997-2011. doi: 10.1128/MCB.26.5.1997-2011.2006. Mol Cell Biol. 2006. PMID: 16479016 Free PMC article.
-
Molecular parameters of hyperthermia for radiosensitization.Crit Rev Eukaryot Gene Expr. 2009;19(3):235-51. doi: 10.1615/critreveukargeneexpr.v19.i3.50. Crit Rev Eukaryot Gene Expr. 2009. PMID: 19883367 Free PMC article. Review.
-
Banking on ATM as a new target in metabolic syndrome.Cell Metab. 2006 Nov;4(5):337-8. doi: 10.1016/j.cmet.2006.10.009. Cell Metab. 2006. PMID: 17084707 Review.
Cited by
-
ATM-dependent ERK signaling via AKT in response to DNA double-strand breaks.Cell Cycle. 2011 Feb 1;10(3):481-91. doi: 10.4161/cc.10.3.14713. Epub 2011 Feb 1. Cell Cycle. 2011. PMID: 21263216 Free PMC article.
-
DIM (3,3'-diindolylmethane) confers protection against ionizing radiation by a unique mechanism.Proc Natl Acad Sci U S A. 2013 Nov 12;110(46):18650-5. doi: 10.1073/pnas.1308206110. Epub 2013 Oct 14. Proc Natl Acad Sci U S A. 2013. PMID: 24127581 Free PMC article.
-
Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies.Cancers (Basel). 2021 Jul 29;13(15):3819. doi: 10.3390/cancers13153819. Cancers (Basel). 2021. PMID: 34359720 Free PMC article. Review.
-
Redox regulation of SUMO enzymes is required for ATM activity and survival in oxidative stress.EMBO J. 2016 Jun 15;35(12):1312-29. doi: 10.15252/embj.201593404. Epub 2016 May 12. EMBO J. 2016. PMID: 27174643 Free PMC article.
-
ATM function and its relationship with ATM gene mutations in chronic lymphocytic leukemia with the recurrent deletion (11q22.3-23.2).Blood Cancer J. 2016 Sep 2;6(9):e465. doi: 10.1038/bcj.2016.69. Blood Cancer J. 2016. PMID: 27588518 Free PMC article.
References
-
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 - PubMed
-
- Bakkenist CJ, Kastan MB (2004) Phosphatases join kinases in DNA-damage response pathways. Trends Cell Biol 14: 339–341 - PubMed
-
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281: 1674–1677 - PubMed
-
- Borthwick EB, Zeke T, Prescott AR, Cohen PT (2001) Nuclear localization of protein phosphatase 5 is dependent on the carboxy-terminal region. FEBS Lett 491: 279–284 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
