

A novel, high-performance random array platform for quantitative gene expression profiling
- PMID: 15520296
- PMCID: PMC525694
- DOI: 10.1101/gr.2739104
A novel, high-performance random array platform for quantitative gene expression profiling
Abstract
We have developed a new microarray technology for quantitative gene-expression profiling on the basis of randomly assembled arrays of beads. Each bead carries a gene-specific probe sequence. There are multiple copies of each sequence-specific bead in an array, which contributes to measurement precision and reliability. We optimized the system for specific and sensitive analysis of mammalian RNA, and using RNA controls of defined concentration, obtained the following estimates of system performance: specificity of 1:250,000 in mammalian poly(A(+)) mRNA; limit of detection 0.13 pM; dynamic range 3.2 logs; and sufficient precision to detect 1.3-fold differences with 95% confidence within the dynamic range. Measurements of expression differences between human brain and liver were validated by concordance with quantitative real-time PCR (R(2) = 0.98 for log-transformed ratios, and slope of the best-fit line = 1.04, for 20 genes). Quantitative performance was further verified using a mouse B- and T-cell model system. We found published reports of B- or T-cell-specific expression for 42 of 59 genes that showed the greatest differential expression between B- and T-cells in our system. All of the literature observations were concordant with our results. Our experiments were carried out on a 96-array matrix system that requires only 100 ng of input RNA and uses standard microtiter plates to process samples in parallel. Our technology has advantages for analyzing multiple samples, is scalable to all known genes in a genome, and is flexible, allowing the use of standard or custom probes in an array.
Figures



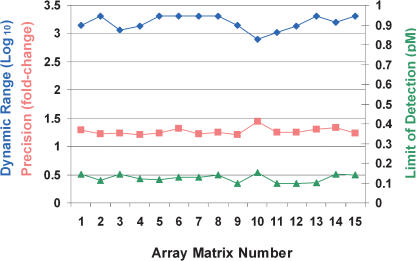

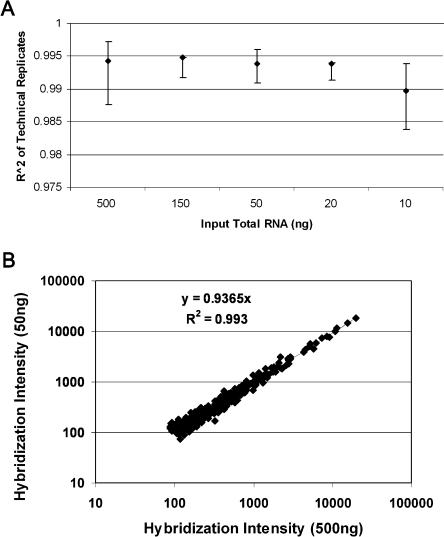


References
-
- Abbas, A.K., Lichtman, A.H., and Pober, J.S. 2003. Cellular and molecular immunology. W.B. Saunders, Philadelphia, PA.
-
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. - PubMed
-
- Barker, D.L., Therault, G., Che, D., Dickinson, T., Shen, R., and Kain, R. 2003. Self-assembled random arrays: High-performance imaging and genomics applications on a high-density microarray platform. Proc. SPIE 4966: 1-11.
-
- Blanchard, A. 1998. Synthetic DNA arrays. Plenum Press, New York.
WEB SITE REFERENCES
-
- www.hapmap.org; International HapMap Project.
-
- www.illumina.com; Illumina, Inc.
-
- www.mged.org; Microarray Gene Expression Data Society.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources