

Mycobacterium tuberculosis functional network analysis by global subcellular protein profiling
- PMID: 15525680
- PMCID: PMC539182
- DOI: 10.1091/mbc.e04-04-0329
Mycobacterium tuberculosis functional network analysis by global subcellular protein profiling
Abstract
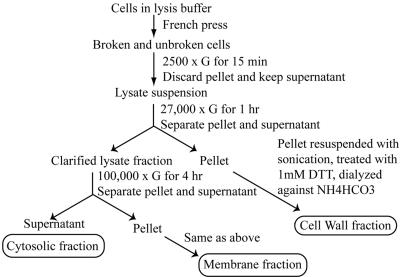
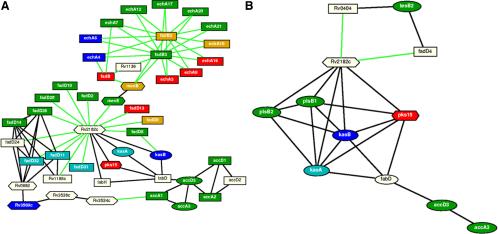
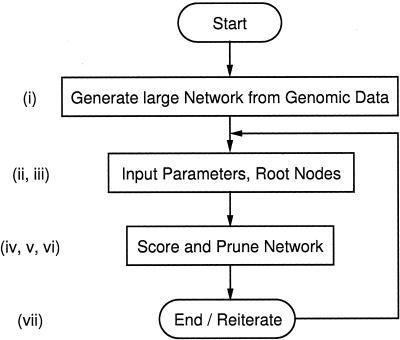
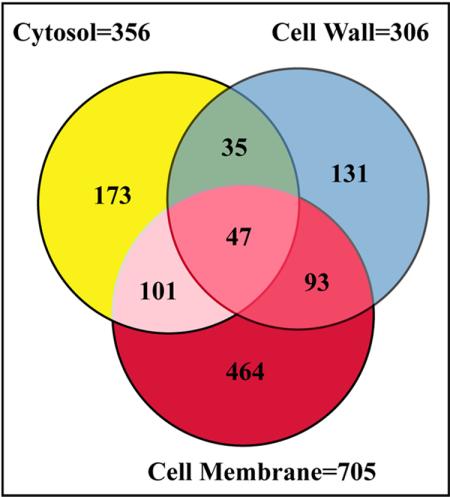
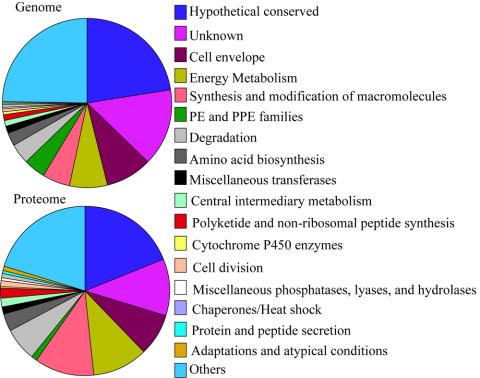
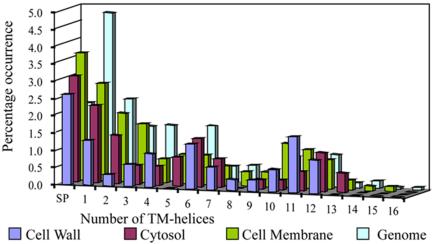
Trends in increased tuberculosis infection and a fatality rate of approximately 23% have necessitated the search for alternative biomarkers using newly developed postgenomic approaches. Here we provide a systematic analysis of Mycobacterium tuberculosis (Mtb) by directly profiling its gene products. This analysis combines high-throughput proteomics and computational approaches to elucidate the globally expressed complements of the three subcellular compartments (the cell wall, membrane, and cytosol) of Mtb. We report the identifications of 1044 proteins and their corresponding localizations in these compartments. Genome-based computational and metabolic pathways analyses were performed and integrated with proteomics data to reconstruct response networks. From the reconstructed response networks for fatty acid degradation and lipid biosynthesis pathways in Mtb, we identified proteins whose involvements in these pathways were not previously suspected. Furthermore, the subcellular localizations of these expressed proteins provide interesting insights into the compartmentalization of these pathways, which appear to traverse from cell wall to cytoplasm. Results of this large-scale subcellular proteome profile of Mtb have confirmed and validated the computational network hypothesis that functionally related proteins work together in larger organizational structures.
Figures
References
-
- Barry, C. (2001). Interpreting cell wall `virulence factors' of Mycobacterium tuberculosis. Trends Microbiol. 9, 237-241. - PubMed
-
- Barry, C., Lee, R., Mdluli, K., Sampson, A., Schroeder, B., Slayden, R., and Yuan, Y. (1998). Mycolic acids: structure, biosynthesis and physiological functions. Prog. Lipid Res. 37, 143-179. - PubMed
-
- Brennan, P., and Nikaido, H. (1995). The envelope of mycobacteria. Annu. Rev. Biochem. 64, 29-63. - PubMed
-
- Camus, J. C., Pryor, M. J., Medigue, C., and Cole, S. T. (2002). Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology 148, 2967-2973. - PubMed
-
- Cole, S. et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete. Nature 393, 537-544. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
