

Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death
- PMID: 15525775
- PMCID: PMC6730239
- DOI: 10.1523/JNEUROSCI.1713-04.2004
Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death
Abstract
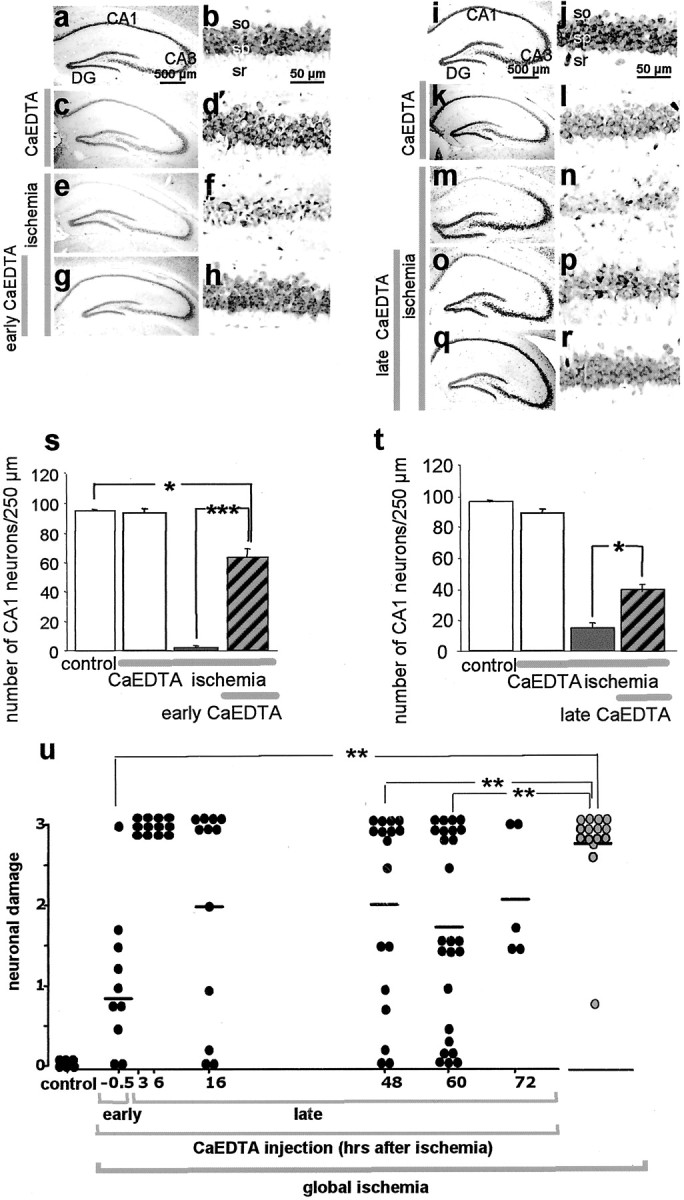
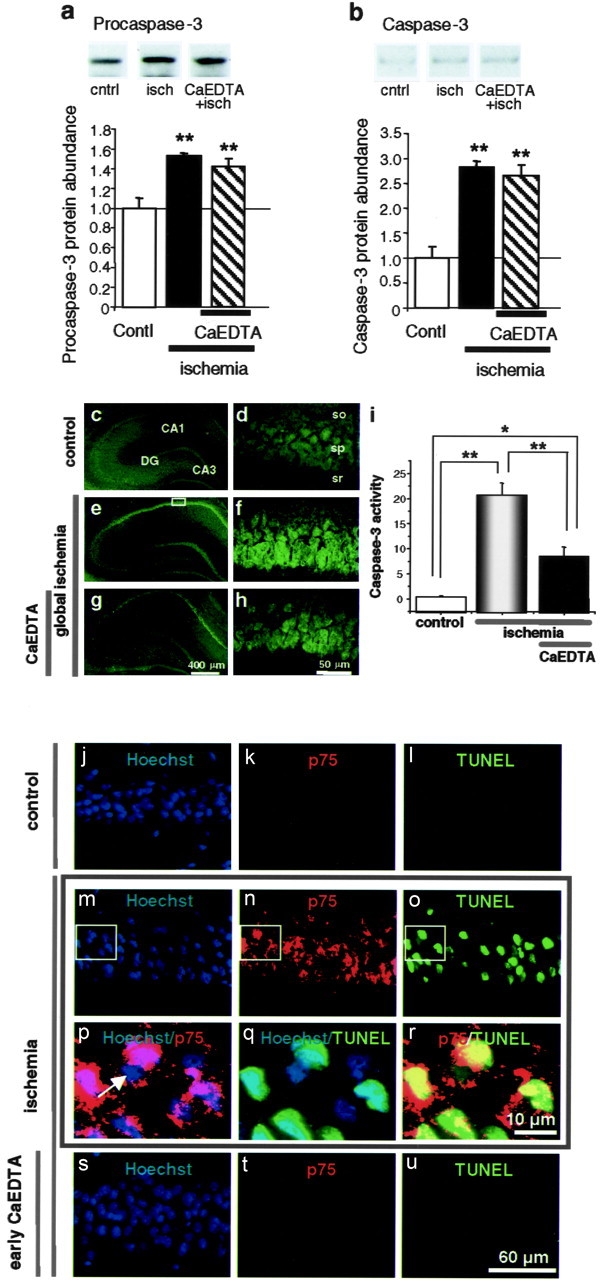
Transient global ischemia induces a delayed rise in intracellular Zn2+, which may be mediated via glutamate receptor 2 (GluR2)-lacking AMPA receptors (AMPARs), and selective, delayed death of hippocampal CA1 neurons. The molecular mechanisms underlying Zn2+ toxicity in vivo are not well delineated. Here we show the striking finding that intraventricular injection of the high-affinity Zn2+ chelator calcium EDTA (CaEDTA) at 30 min before ischemia (early CaEDTA) or at 48-60 hr (late CaEDTA), but not 3-6 hr, after ischemia, afforded robust protection of CA1 neurons in approximately 50% (late CaEDTA) to 75% (early CaEDTA) of animals. We also show that Zn2+ acts via temporally distinct mechanisms to promote neuronal death. Early CaEDTA attenuated ischemia-induced GluR2 mRNA and protein downregulation (and, by inference, formation of Zn2+-permeable AMPARs), the delayed rise in Zn2+, and neuronal death. These findings suggest that Zn2+ acts at step(s) upstream from GluR2 gene downregulation and implicate Zn2+ in transcriptional regulation and/or GluR2 mRNA stability. Early CaEDTA also blocked mitochondrial release of cytochrome c and Smac/DIABLO (second mitochondria-derived activator of caspases/direct inhibitor of apoptosis protein-binding protein with low pI), caspase-3 activity (but not procaspase-3 cleavage), p75NTR induction, and DNA fragmentation. These findings indicate that CaEDTA preserves the functional integrity of the mitochondrial outer membrane and arrests the caspase death cascade. Late injection of CaEDTA at a time when GluR2 is downregulated and caspase is activated inhibited the delayed rise in Zn2+, p75NTR induction, DNA fragmentation, and cell death. The finding of neuroprotection by late CaEDTA administration has striking implications for intervention in the delayed neuronal death associated with global ischemia.
Figures




Similar articles
-
Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death.Proc Natl Acad Sci U S A. 2005 Aug 23;102(34):12230-5. doi: 10.1073/pnas.0505408102. Epub 2005 Aug 10. Proc Natl Acad Sci U S A. 2005. PMID: 16093311 Free PMC article.
-
Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1.J Neurosci. 2002 Mar 15;22(6):2115-24. doi: 10.1523/JNEUROSCI.22-06-02115.2002. J Neurosci. 2002. PMID: 11896151 Free PMC article.
-
Ischemic preconditioning: neuronal survival in the face of caspase-3 activation.J Neurosci. 2004 Mar 17;24(11):2750-9. doi: 10.1523/JNEUROSCI.5475-03.2004. J Neurosci. 2004. PMID: 15028768 Free PMC article.
-
The AMPAR subunit GluR2: still front and center-stage.Brain Res. 2000 Dec 15;886(1-2):190-207. doi: 10.1016/s0006-8993(00)02951-6. Brain Res. 2000. PMID: 11119696 Review.
-
Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra.J Neuropathol Exp Neurol. 2003 Apr;62(4):329-39. doi: 10.1093/jnen/62.4.329. J Neuropathol Exp Neurol. 2003. PMID: 12722825 Review.
Cited by
-
Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain.J Neurosci. 2006 Jun 21;26(25):6851-62. doi: 10.1523/JNEUROSCI.5444-05.2006. J Neurosci. 2006. PMID: 16793892 Free PMC article.
-
Spreading depression and related events are significant sources of neuronal Zn2+ release and accumulation.J Cereb Blood Flow Metab. 2011 Apr;31(4):1073-84. doi: 10.1038/jcbfm.2010.183. Epub 2010 Oct 27. J Cereb Blood Flow Metab. 2011. PMID: 20978516 Free PMC article.
-
MAPK signaling is critical to estradiol protection of CA1 neurons in global ischemia.Endocrinology. 2007 Mar;148(3):1131-43. doi: 10.1210/en.2006-1137. Epub 2006 Nov 30. Endocrinology. 2007. PMID: 17138646 Free PMC article.
-
Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System.Front Neurosci. 2018 Apr 20;12:214. doi: 10.3389/fnins.2018.00214. eCollection 2018. Front Neurosci. 2018. PMID: 29731704 Free PMC article. Review.
-
Zinc inhibits astrocyte glutamate uptake by activation of poly(ADP-ribose) polymerase-1.Mol Med. 2007 Jul-Aug;13(7-8):344-9. doi: 10.2119/2007-00043.Suh. Mol Med. 2007. PMID: 17728843 Free PMC article.
References
-
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ (2000) Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem 75: 1878-1888. - PubMed
-
- Bers DM, Patton CW, Nuccitelli R (1994) A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol 40: 3-29. - PubMed
-
- Brene S, Messer C, Okado H, Hartley M, Heinemann SF, Nestler EJ (2000) Regulation of GluR2 promoter activity by neurotrophic factors via a neuron-restrictive silencer element. Eur J Neurosci 12: 1525-1533. - PubMed
-
- Bresink I, Ebert B, Parsons CG, Mutschler E (1996) Zinc changes AMPA receptor properties: results of binding studies and patch clamp recordings. Neuropharmacology 35: 503-509. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous