

Retinoids arrest breast cancer cell proliferation: retinoic acid selectively reduces the duration of receptor tyrosine kinase signaling
- PMID: 15530851
- PMCID: PMC2742418
- DOI: 10.1016/j.yexcr.2004.07.008
Retinoids arrest breast cancer cell proliferation: retinoic acid selectively reduces the duration of receptor tyrosine kinase signaling
Abstract
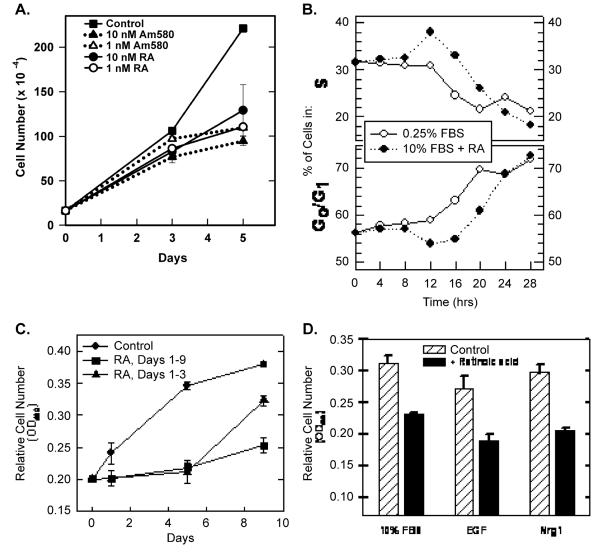
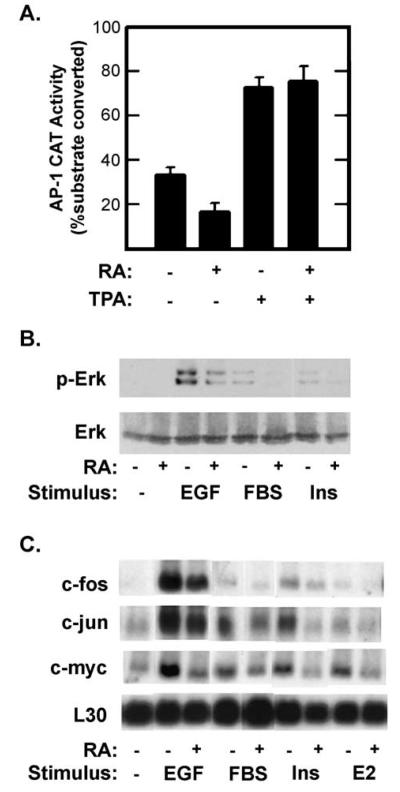
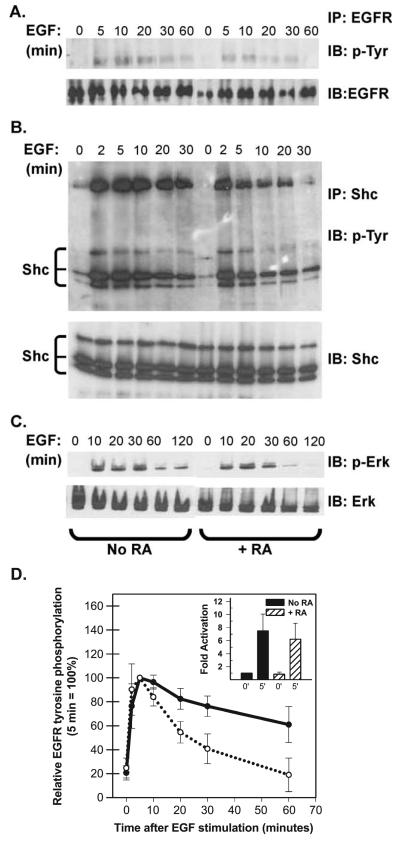
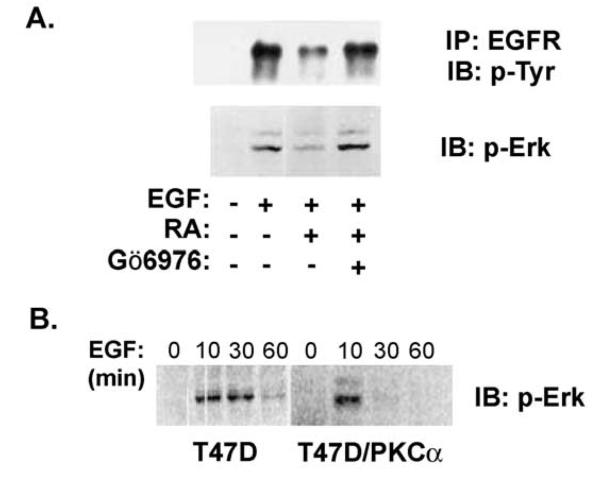
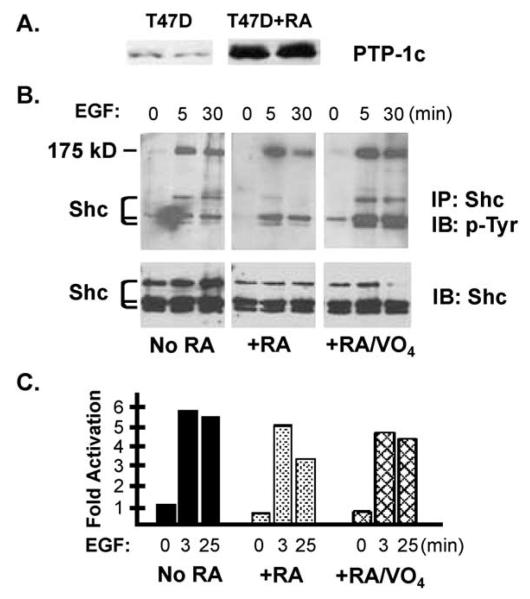
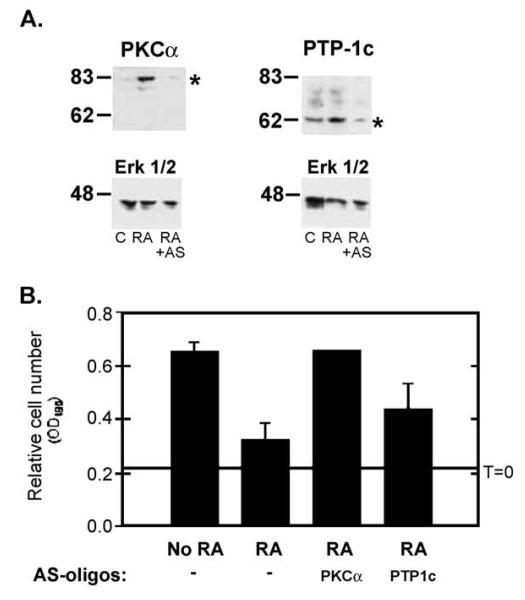
Retinoic acid (RA) induces cell cycle arrest of hormone-dependent human breast cancer (HBC) cells. Previously, we demonstrated that RA-induced growth arrest of T-47D HBC cells required the activity of the RA-induced protein kinase, protein kinase Calpha (PKCalpha) [J. Cell Physiol. 172 (1997) 306]. Here, we demonstrate that RA treatment of T-47D cells interfered with growth factor signaling to downstream, cytoplasmic and nuclear targets. RA treatment did not inhibit epidermal growth factor (EGF) receptor activation but resulted in rapid inactivation. The lack of sustained EGFR activation was associated with transient rather than sustained association of the EGFR with the Shc adaptor proteins and activation of Erk 1/2 and with compromised induction of expression of immediate early response genes. Inhibiting the activity of PKCalpha, a retinoic acid-induced target gene, prevented the effects of RA on cell proliferation and EGF signaling. Constitutive expression of PKCalpha, in the absence of RA, decreased cell proliferation and decreased EGF signaling. RA treatment increased steady-state levels of the protein tyrosine phosphatase PTP-1C and all measured effects of RA on EGF receptor function were reversed by the tyrosine phosphate inhibitor orthovanadate. These results indicate that RA-induced target genes, particularly PKCalpha, prevent sustained growth factor signaling, uncoupling activated receptor tyrosine kinases and nuclear targets that are required for cell cycle progression.
Figures
References
-
- Cho Y, Tighe AP, Talmage DA. Retinoic acid induced growth arrest of human breast carcinoma cells requires protein kinase C alpha expression and activity. J. Cell. Physiol. 1997;172:306–313. - PubMed
-
- Lotan R. Retinoids in cancer chemoprevention. FASEB J. 1996;10:1031–1039. - PubMed
-
- Cho Y, Talmage DA. Protein kinase C-alpha expression confers retinoic acid sensitivity on MDA-MB-231 human breast cancer cells. Exp. Cell Res. 2001;269:97–108. - PubMed
-
- Dow R, Hendley J, Pirkmaier A, Musgrove EA, Germain D. Retinoic acid-mediated growth arrest requires ubiquitylation and degradation of the F-box protein Skp2. J. Biol. Chem. 2001;276:45945–45951. - PubMed
-
- Fitzgerald P, Teng M, Chandraratna RA, Heyman RA, Allegretto EA. Retinoic acid receptor alpha expression correlates with retinoid-induced growth inhibition of human breast cancer cells regardless of estrogen receptor status. Cancer Res. 1997;57:2642–2650. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
