

Experimental validation of molecular dynamics simulations of lipid bilayers: a new approach
- PMID: 15533925
- PMCID: PMC1305157
- DOI: 10.1529/biophysj.104.046821
Experimental validation of molecular dynamics simulations of lipid bilayers: a new approach
Abstract
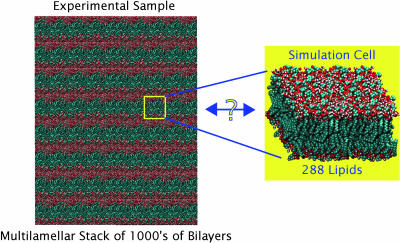
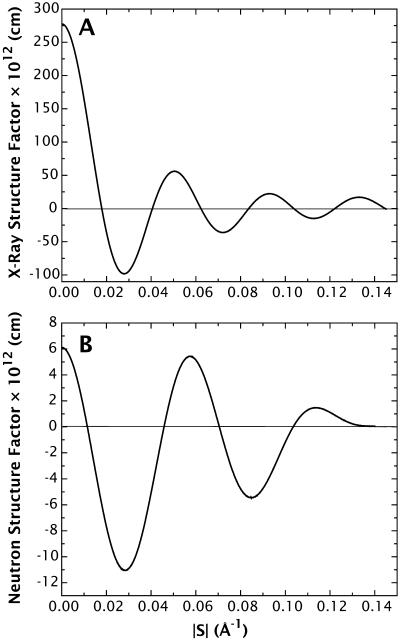
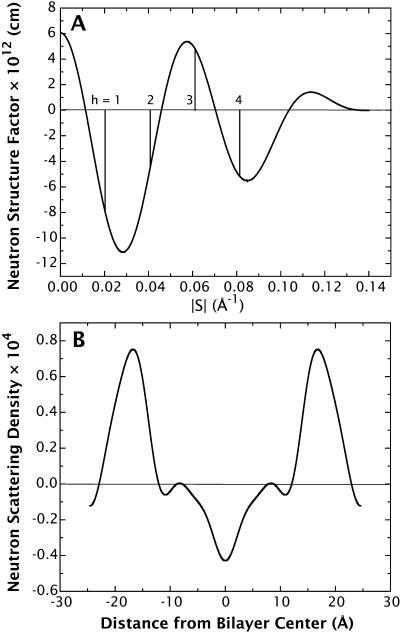
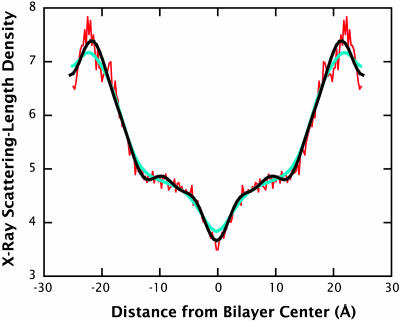
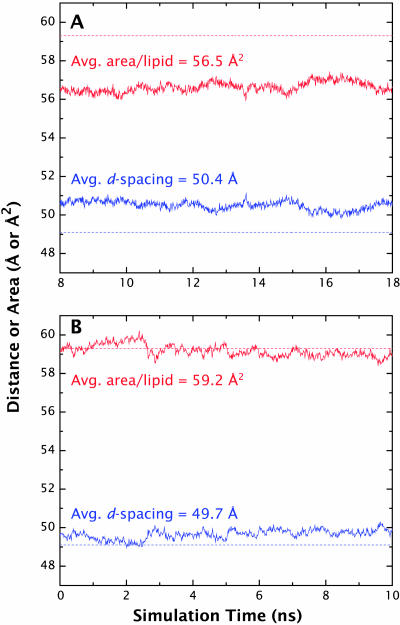
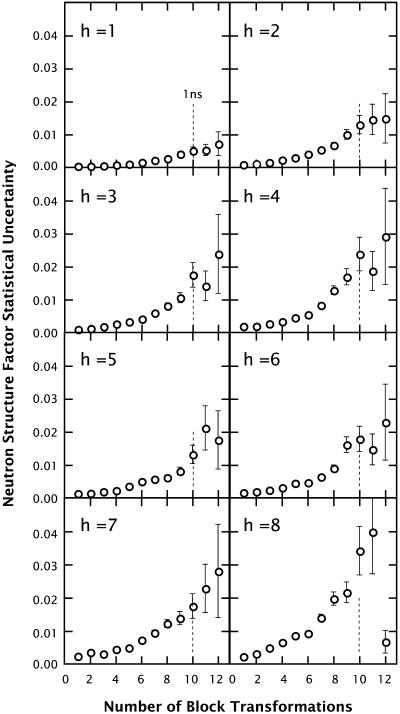
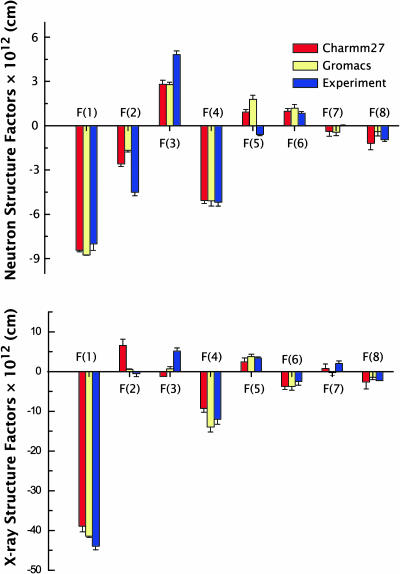
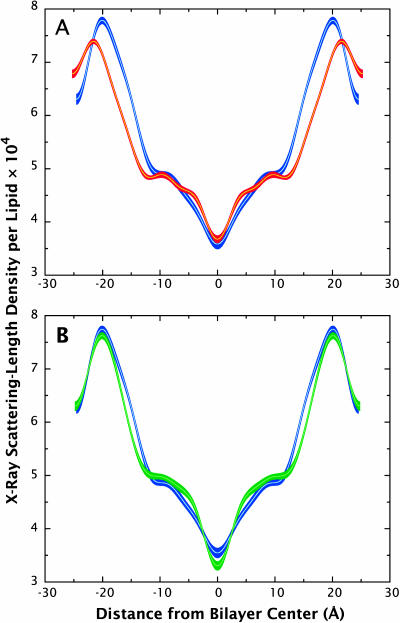
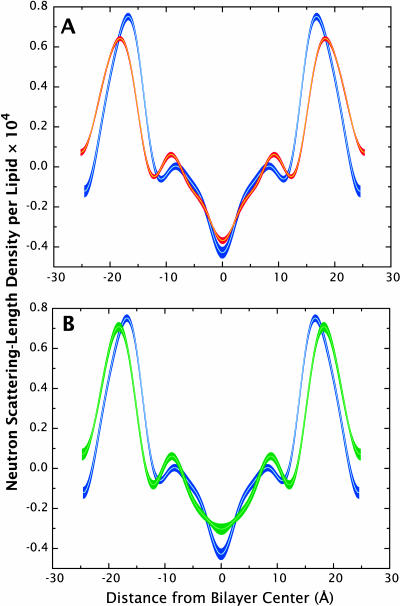
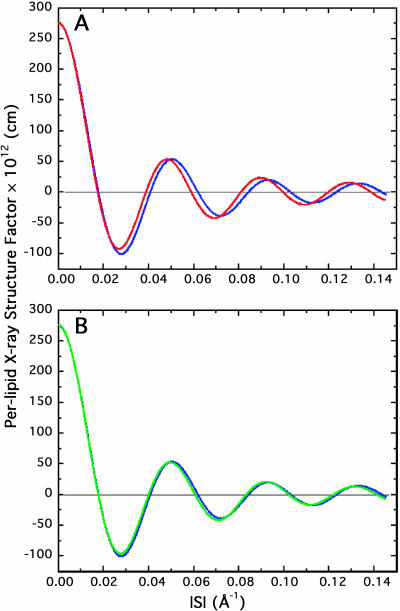
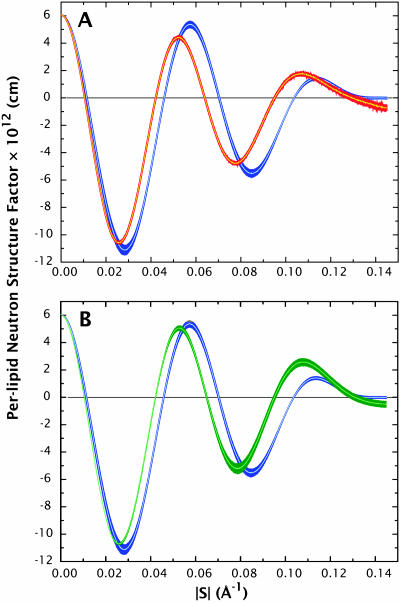
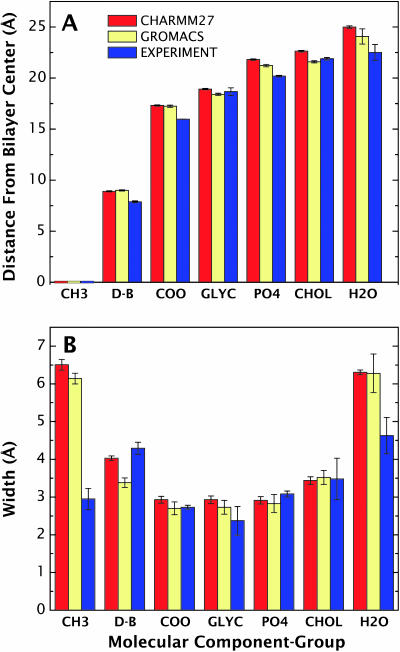
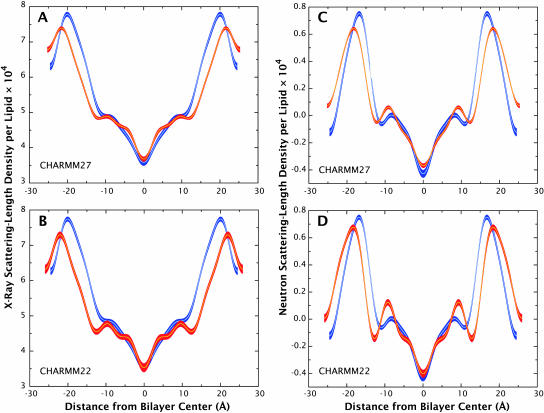
A novel protocol has been developed for comparing the structural properties of lipid bilayers determined by simulation with those determined by diffraction experiments, which makes it possible to test critically the ability of molecular dynamics simulations to reproduce experimental data. This model-independent method consists of analyzing data from molecular dynamics bilayer simulations in the same way as experimental data by determining the structure factors of the system and, via Fourier reconstruction, the overall transbilayer scattering-density profiles. Multi-nanosecond molecular dynamics simulations of a dioleoylphosphatidylcholine bilayer at 66% RH (5.4 waters/lipid) were performed in the constant pressure and temperature ensemble using the united-atom GROMACS and the all-atom CHARMM22/27 force fields with the GROMACS and NAMD software packages, respectively. The quality of the simulated bilayer structures was evaluated by comparing simulation with experimental results for bilayer thickness, area/lipid, individual molecular-component distributions, continuous and discrete structure factors, and overall scattering-density profiles. Neither the GROMACS nor the CHARMM22/27 simulations reproduced experimental data within experimental error. The widths of the simulated terminal methyl distributions showed a particularly strong disagreement with the experimentally observed distributions. A comparison of the older CHARMM22 with the newer CHARMM27 force fields shows that significant progress is being made in the development of atomic force fields for describing lipid bilayer systems empirically.
Figures
References
-
- Allen, M. P., and D. J. Tildesley. 1987. Computer Simulation of Liquids. Oxford University Press, Oxford, UK.
-
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690.
-
- Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a new message-passing parallel molecular dynamics implementation. Comp. Phys. Commun. 91:43–56.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
