

Conservation and evolution of cis-regulatory systems in ascomycete fungi
- PMID: 15534694
- PMCID: PMC526180
- DOI: 10.1371/journal.pbio.0020398
Conservation and evolution of cis-regulatory systems in ascomycete fungi
Abstract
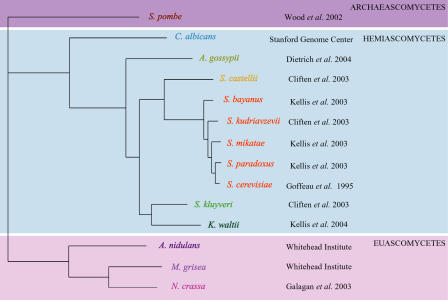
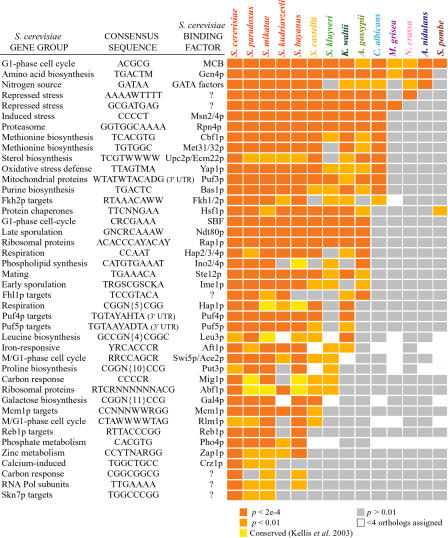
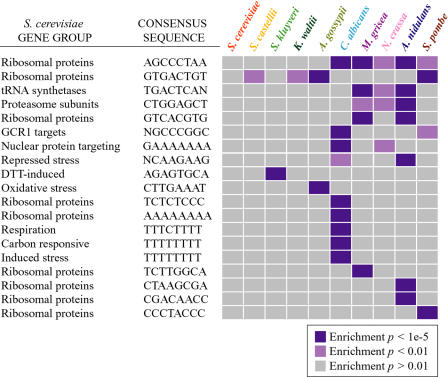
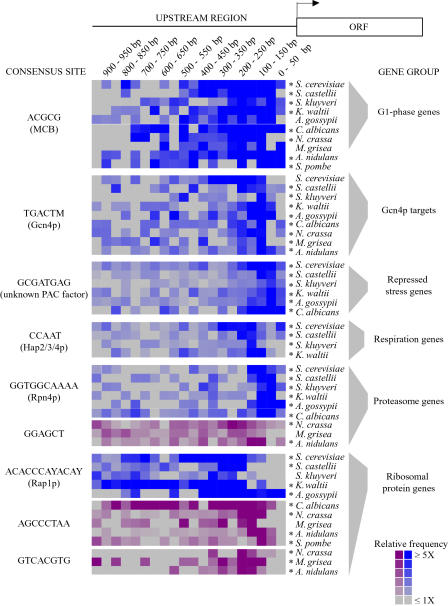
Relatively little is known about the mechanisms through which gene expression regulation evolves. To investigate this, we systematically explored the conservation of regulatory networks in fungi by examining the cis-regulatory elements that govern the expression of coregulated genes. We first identified groups of coregulated Saccharomyces cerevisiae genes enriched for genes with known upstream or downstream cis-regulatory sequences. Reasoning that many of these gene groups are coregulated in related species as well, we performed similar analyses on orthologs of coregulated S. cerevisiae genes in 13 other ascomycete species. We find that many species-specific gene groups are enriched for the same flanking regulatory sequences as those found in the orthologous gene groups fromS. cerevisiae, indicating that those regulatory systems have been conserved in multiple ascomycete species. In addition to these clear cases of regulatory conservation, we find examples of cis-element evolution that suggest multiple modes of regulatory diversification, including alterations in transcription factor-binding specificity, incorporation of new gene targets into an existing regulatory system, and cooption of regulatory systems to control a different set of genes. We investigated one example in greater detail by measuring the in vitro activity of the S. cerevisiae transcription factor Rpn4p and its orthologs from Candida albicans and Neurospora crassa. Our results suggest that the DNA binding specificity of these proteins has coevolved with the sequences found upstream of the Rpn4p target genes and suggest that Rpn4p has a different function in N. crassa.
Conflict of interest statement
The authors have declared that no conflicts of interest exist.
Figures
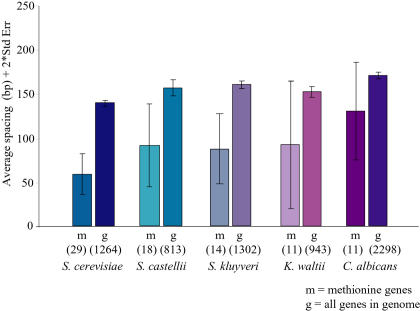

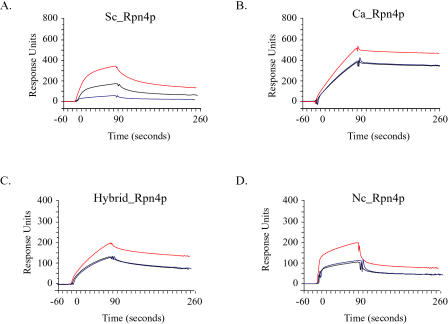
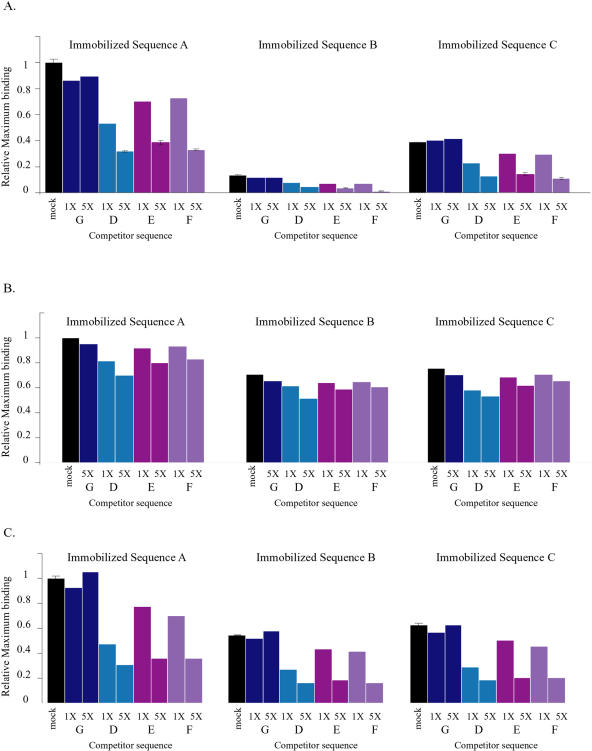

References
-
- Ainsworth GC, Kirk PM, Bisby GR. Dictionary of the fungi. Kirk PM, Cannon PF, David JC, editors. Egham, UK: CABI Publishing; 2001. 616 p pp.
-
- Averof M, Patel NH. Crustacean appendage evolution associated with changes in Hox gene expression. Nature. 1997;388:682–686. - PubMed
-
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. - PubMed
-
- Benos PV, Lapedes AS, Stormo GD. Probabilistic code for DNA recognition by proteins of the EGR family. J Mol Biol. 2002;323:701–727. - PubMed
-
- Berbee ML, Taylor JW. Dating the evolutionary radiations of the true fungi. Can J Bot. 1993;71:1114–1127.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
