

Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans
- PMID: 15537665
- PMCID: PMC2858222
- DOI: 10.1093/hmg/ddi010
Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans
Abstract
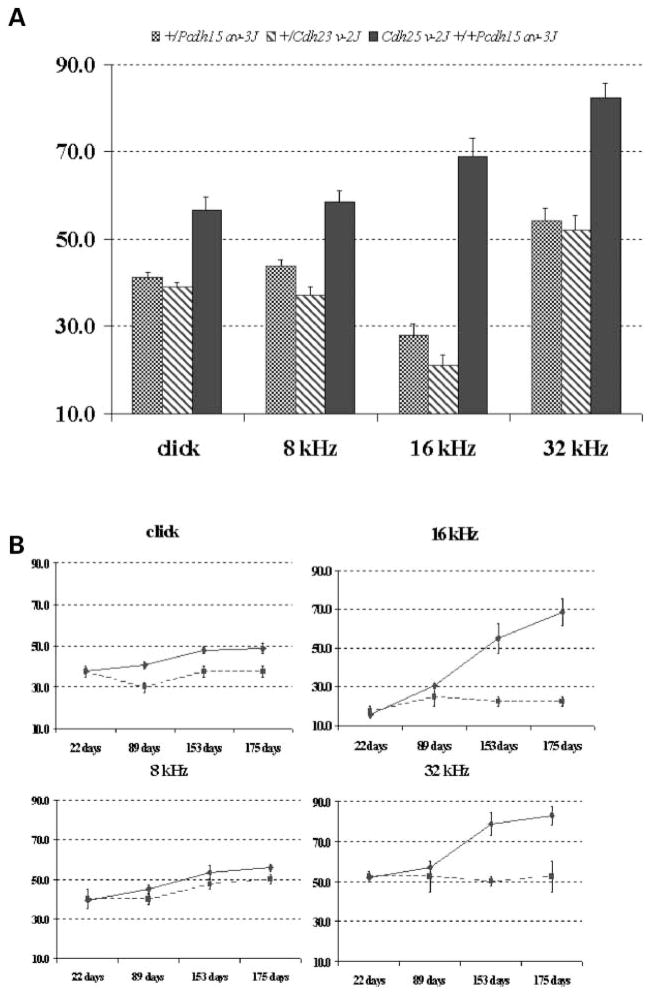
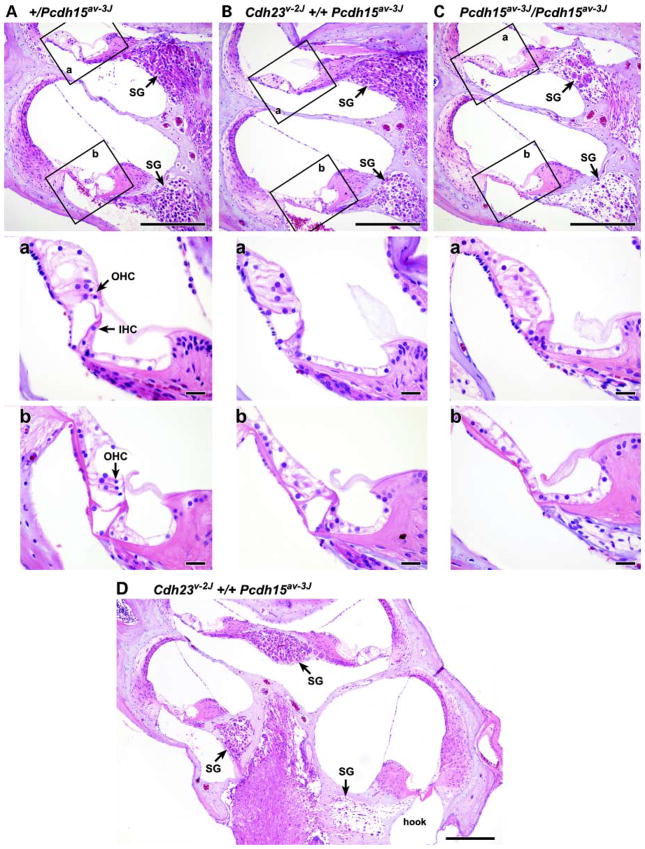
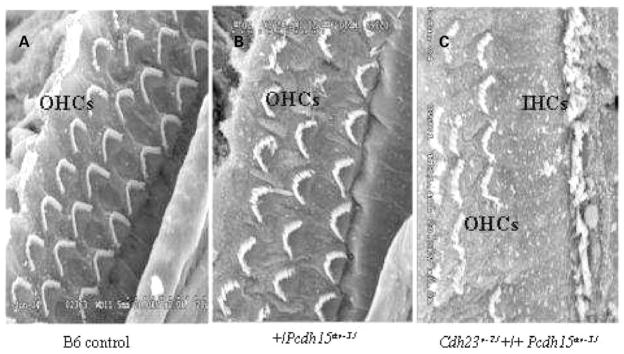
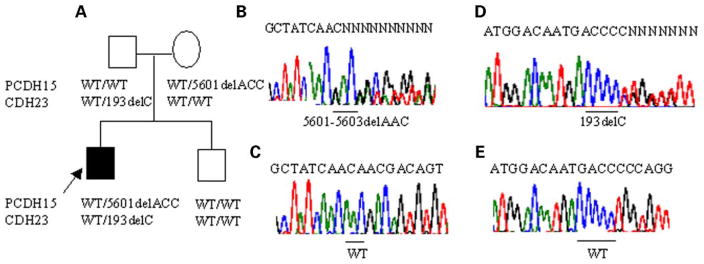
Mutations in genes coding for cadherin 23 and protocadherin 15 cause deafness in both mice and humans. Here, we provide evidence that mutations at these two cadherin loci can interact to cause hearing loss in digenic heterozygotes of both species. Using a classical genetic approach, we generated mice that were heterozygous for both Cdh23 and Pcdh15 mutations on a uniform C57BL/6J background. Significant levels of hearing loss were detected in these mice when compared to age-matched single heterozygous animals or normal controls. Cytoarchitectural defects in the cochlea of digenic heterozygotes, including degeneration of the stereocilia and a base-apex loss of hair cells and spiral ganglion cells, were consistent with the observed age-related hearing loss of these mice beginning with the high frequencies. In humans, we also have obtained evidence for a digenic inheritance of a USH1 phenotype in three unrelated families with mutations in CDH23 and PCDH15. Altogether, our data indicate that CDH23 and PCDH15 play an essential long-term role in maintaining the normal organization of the stereocilia bundle.
Figures
References
-
- Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, del CSCM, Vila MC, Molina OP, et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome typ.1D. Nat Genet. 2001;27:108–112. - PubMed
-
- Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001;68:26–37. - PMC - PubMed
-
- Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CR, Lowry RB, Knaus R, Van Laer L, Bernier FP, et al. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet. 2001;10:1709–1718. - PubMed
-
- Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Riazuddin S, Griffith AJ, Friedman TB, et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet. 2003;12:3215–3223. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
