

Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR
- PMID: 15542627
- PMCID: PMC524969
- DOI: 10.1128/JVI.78.23.12747-12761.2004
Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR
Abstract
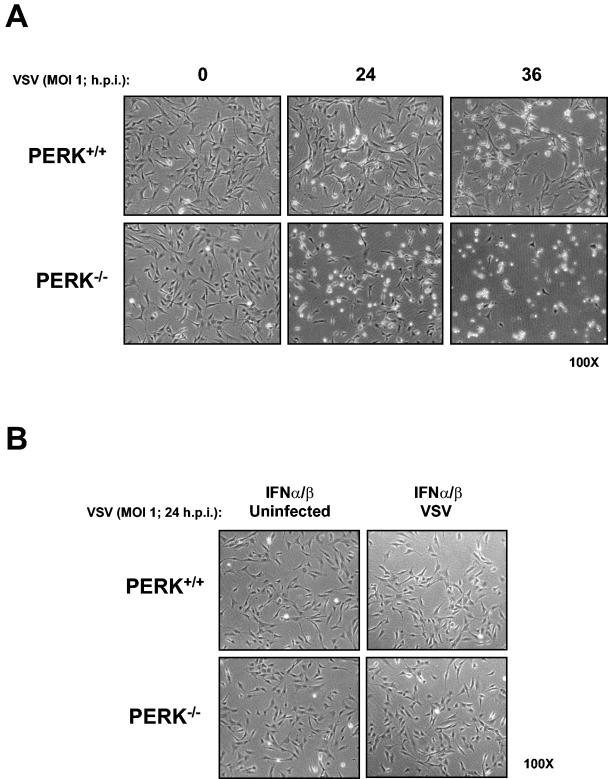
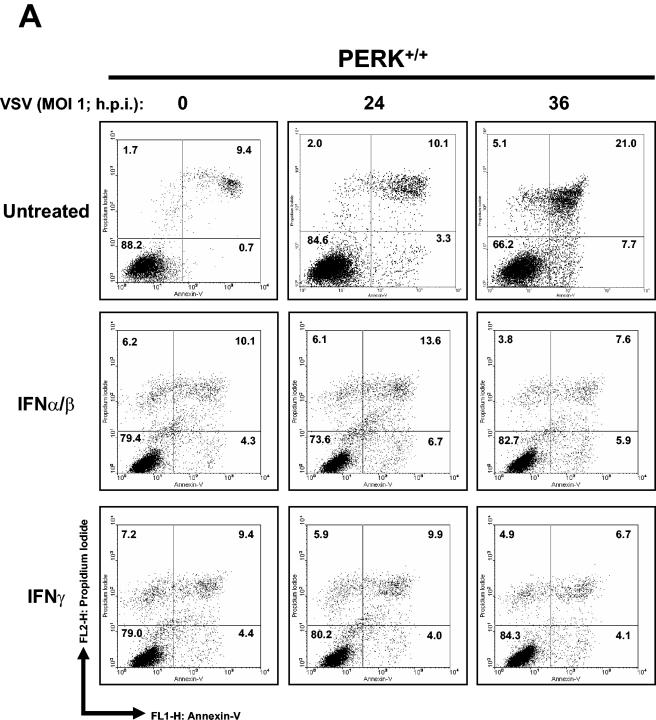
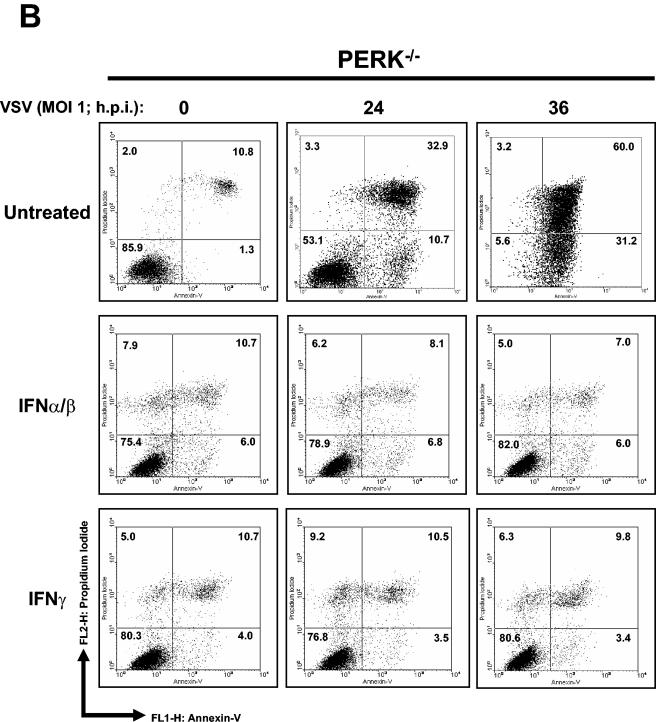
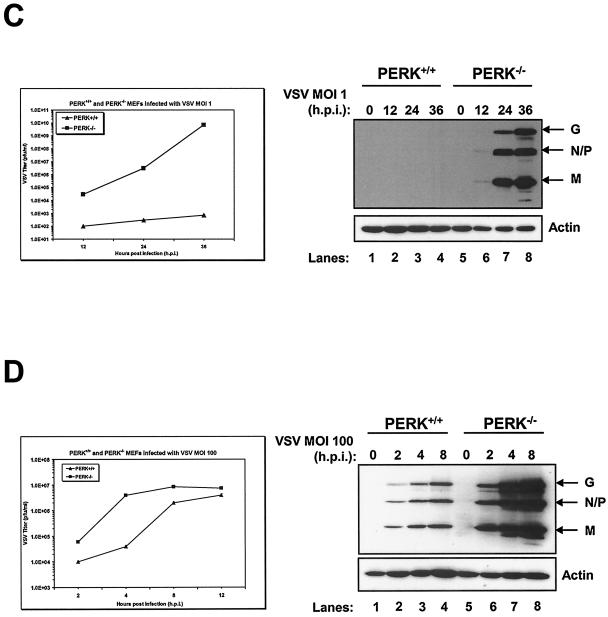
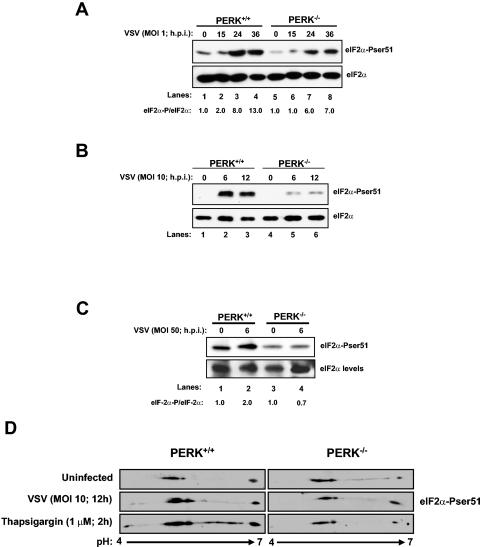
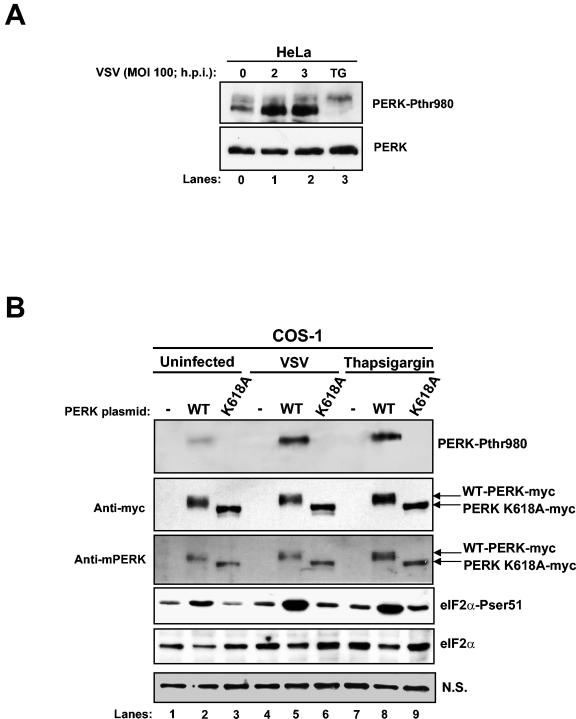
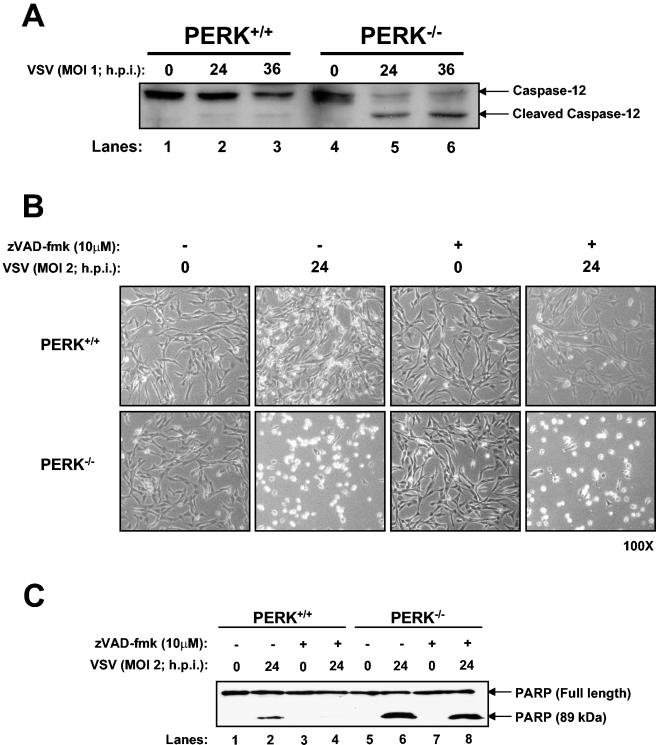
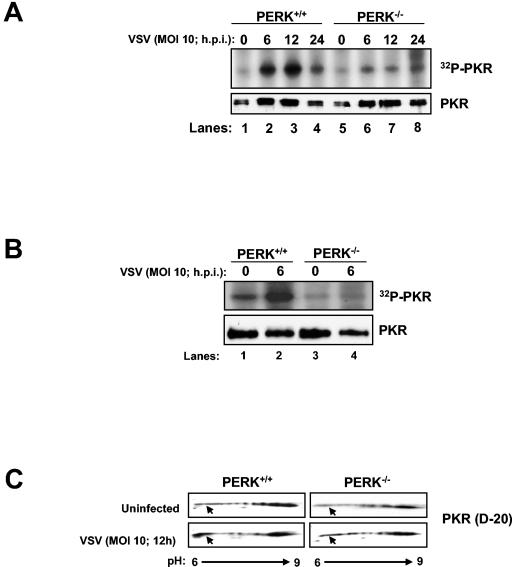
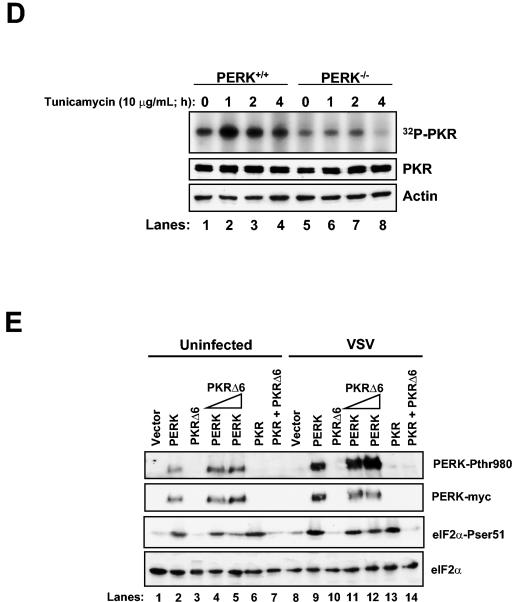
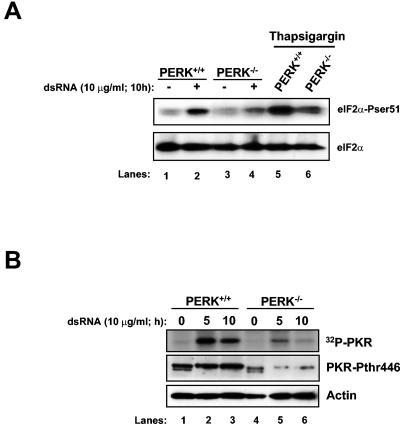
Phosphorylation of the alpha (alpha) subunit of the eukaryotic translation initiation factor 2 (eIF2) leads to the inhibition of protein synthesis in response to diverse stress conditions, including viral infection. The eIF2alpha kinase PKR has been shown to play an essential role against vesicular stomatitis virus (VSV) infection. We demonstrate here that another eIF2alpha kinase, the endoplasmic reticulum-resident protein kinase PERK, contributes to cellular resistance to VSV infection. We demonstrate that mouse embryonic fibroblasts (MEFs) from PERK(-/-) mice are more susceptible to VSV-mediated apoptosis than PERK(+/+) MEFs. The higher replication capacity of VSV in PERK(-/-) MEFs results from their inability to attenuate viral protein synthesis due to an impaired eIF2alpha phosphorylation. We also show that VSV-infected PERK(-/-) MEFs are unable to fully activate PKR, suggesting a cross talk between the two eIF2alpha kinases in virus-infected cells. These findings further implicate PERK in virus infection, and provide evidence that the antiviral and antiapoptotic roles of PERK are mediated, at least in part, via the activation of PKR.
Figures

References
-
- Aridor, M., and W. E. Balch. 1999. Integration of endoplasmic reticulum signaling in health and disease. Nat. Med. 5:745-751. - PubMed
-
- Balachandran, S., P. C. Roberts, L. E. Brown, H. Truong, A. K. Pattnaik, D. R. Archer, and G. N. Barber. 2000. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 13:129-141. - PubMed
-
- Baltzis, D., S. Li, and A. E. Koromilas. 2002. Functional characterization of pkr gene products expressed in cells from mice with a targeted deletion of the N terminus or C terminus domain of PKR. J. Biol. Chem. 277:38364-38372. - PubMed
-
- Bitko, V., and S. Barik. 2001. An endoplasmic reticulum-specific stress-activated caspase (caspase-12) is implicated in the apoptosis of A549 epithelial cells by respiratory syncytial virus. J. Cell Biochem. 80:441-454. - PubMed
-
- Chen, J. J. 2000. Heme-regulated eIF2α kinase, p. 529-546. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
