

p53 Stabilization and accumulation induced by human vaccinia-related kinase 1
- PMID: 15542844
- PMCID: PMC529057
- DOI: 10.1128/MCB.24.23.10366-10380.2004
p53 Stabilization and accumulation induced by human vaccinia-related kinase 1
Abstract
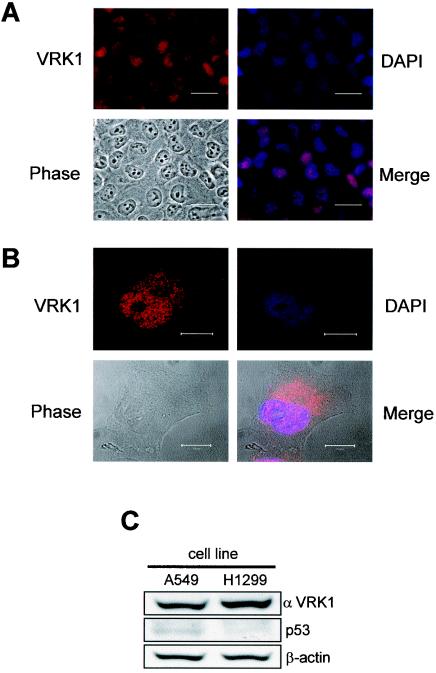
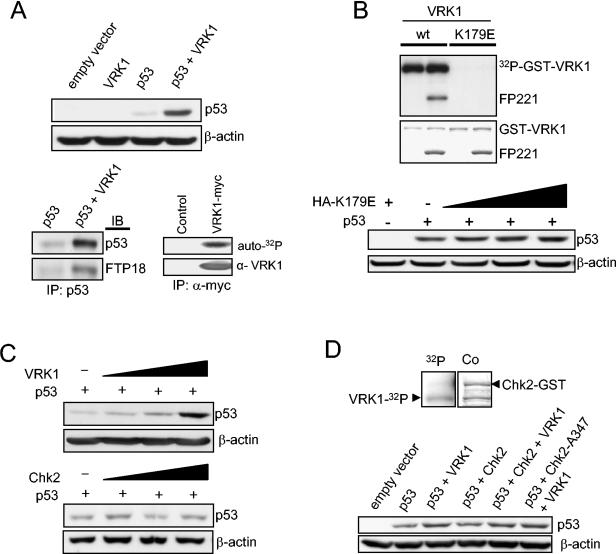
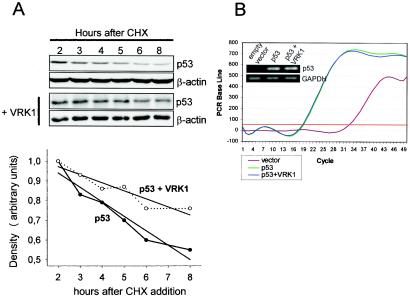
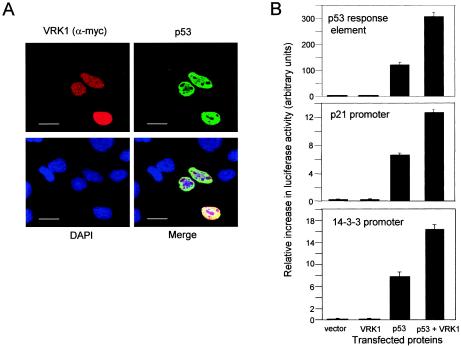
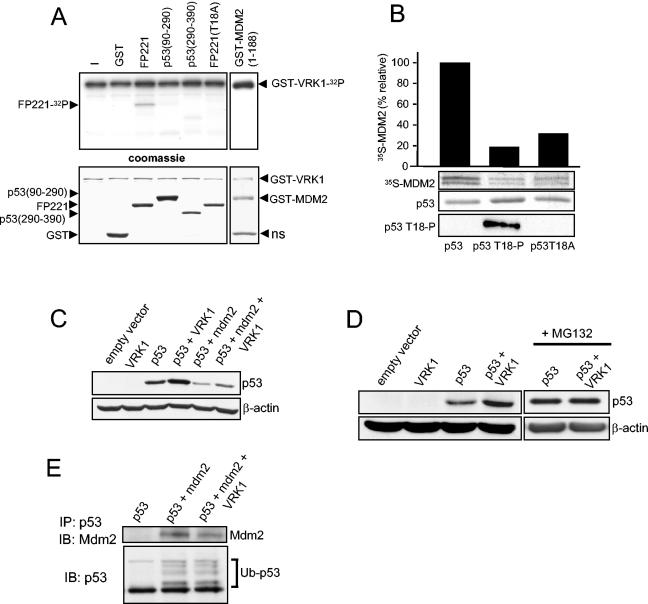
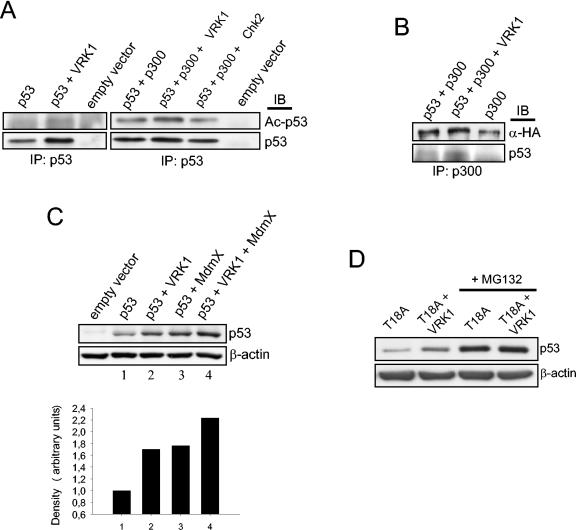
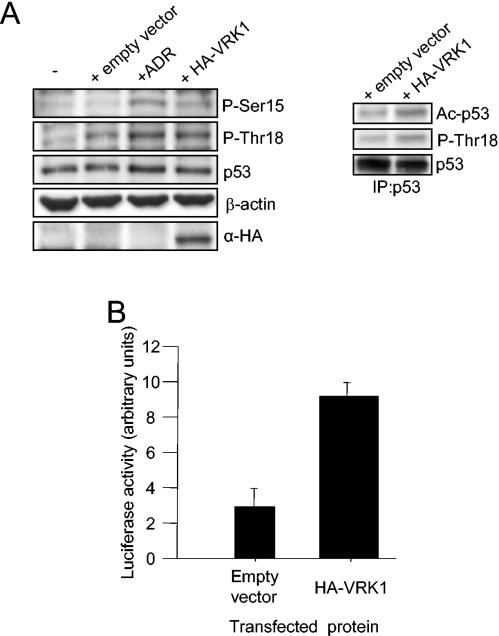
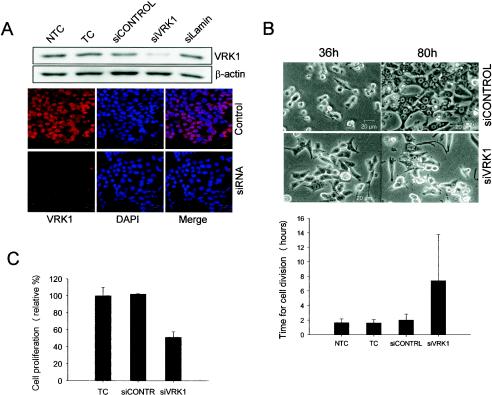
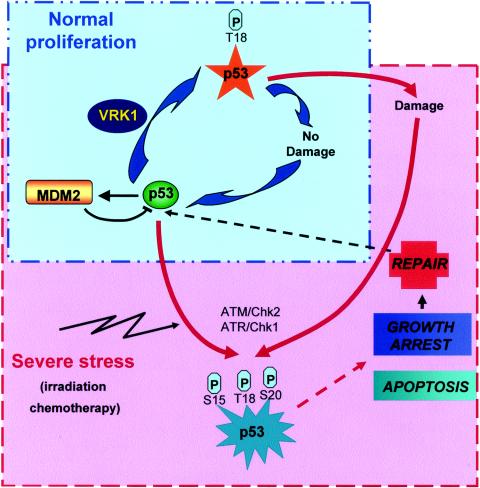
Variations in intracellular levels of p53 regulate many cellular functions and determine tumor susceptibility. Major mechanisms modulating p53 levels include phosphorylation and interaction of p53 with specific ubiquitin ligases that promote its degradation. N-terminal phosphorylation regulates the interaction of p53 with several regulatory molecules. Vaccinia-related kinase 1 (VRK1) is the prototype of a new Ser-Thr kinase family in the human kinome. VRK1 is located in the nucleus outside the nucleolus. Overexpression of VRK1 increases the stability of p53 by a posttranslational mechanism leading to its accumulation by a mechanism independent of the Chk2 kinase. Catalytically inactive VRK1 protein (a K179E mutant) does not induce p53 accumulation. VRK1 phosphorylates human p53 in Thr18 and disrupts p53-Mdm2 interaction in vitro, although a significant decrease in p53 ubiquitination by Mdm2 in vivo was not detected. VRK1 kinase does not phosphorylate Mdm2. VRK1-mediated p53 stabilization was also detected in Mdm2(-/-) cells. VRK1 also has an additive effect with MdmX or p300 to stabilize p53, and p300 coactivation and acetylation of p53 is enhanced by VRK1. The p53 stabilized by VRK1 is transcriptionally active. Suppression of VRK1 expression by specific small interfering RNA provokes several defects in proliferation, situating the protein in the regulation of this process. VRK1 might function as a switch controlling the proteins that interact with p53 and thus modifying its stability and activity. We propose VRK1 as the first step in a new pathway regulating p53 activity during cell proliferation.
Figures
References
-
- Ahn, J., M. Urist, and C. Prives. 2003. Questioning the role of checkpoint kinase 2 in the p53 DNA damage response. J. Biol. Chem. 278:20480-20489. - PubMed
-
- Ashcroft, M., and K. H. Vousden. 1999. Regulation of p53 stability. Oncogene 18:7637-7643. - PubMed
-
- Banham, A. H., and G. L. Smith. 1992. Vaccinia virus B1R encodes a 34-kDa serine/threonine protein kinase that localizes in cytoplasmic factories and is packaged into virions. Virology 191:803-812. - PubMed
-
- Barcia, R., S. Lopez-Borges, F. M. Vega, and P. A. Lazo. 2002. Kinetic properties of p53 phosphorylation by the human vaccinia-related kinase 1. Arch. Biochem. Biophys. 399:1-5. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous