

The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation
- PMID: 15544620
- PMCID: PMC1809239
- DOI: 10.1111/j.1365-2249.2004.02627.x
The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation
Abstract
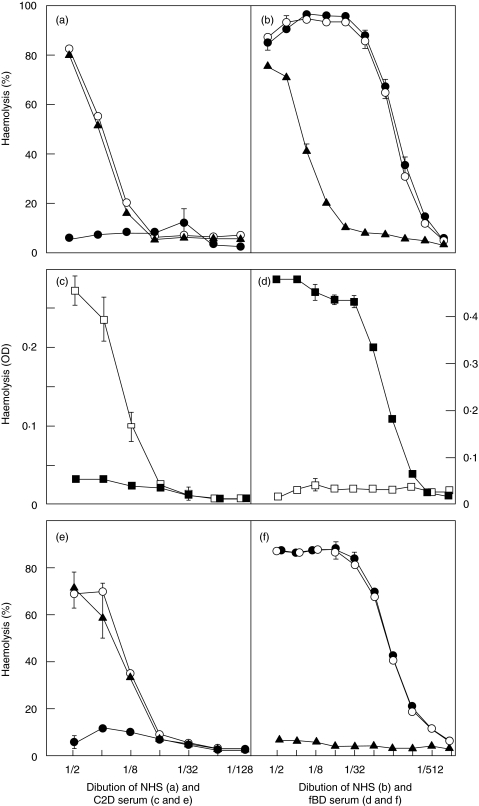
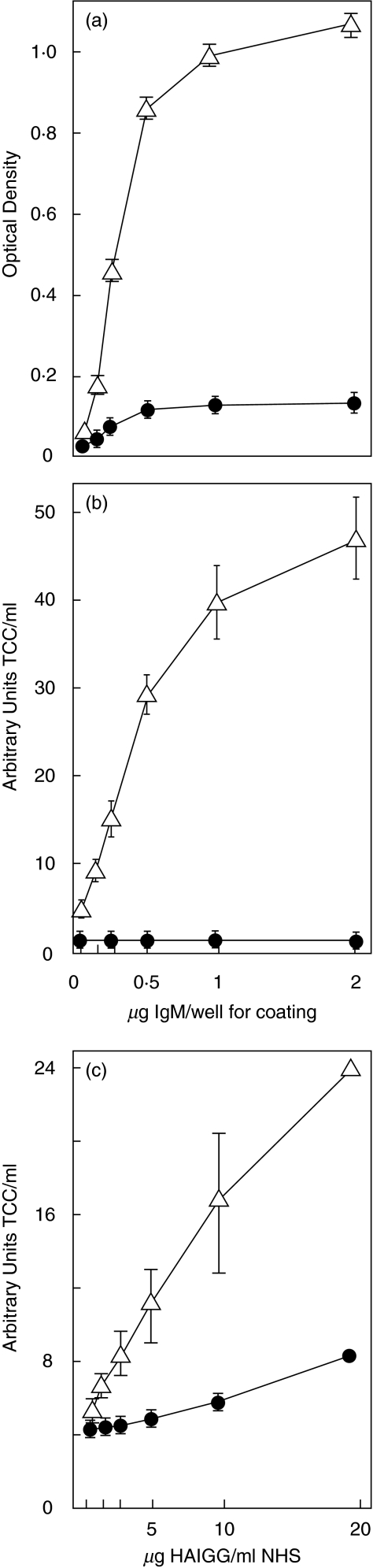
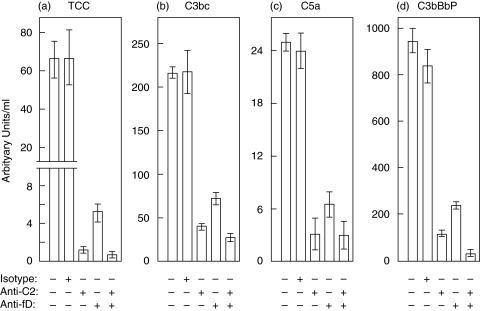
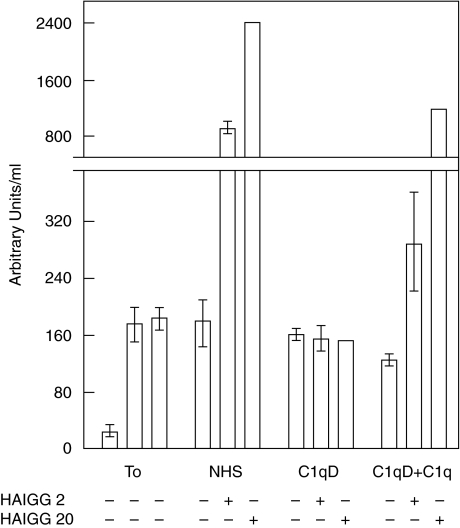
Complement activation with formation of biologically potent mediators like C5a and the terminal C5b-9 complex (TCC) contributes essentially to development of inflammation and tissue damage in a number of autoimmune and inflammatory conditions. A particular role for complement in the ischaemia/reperfusion injury of the heart, skeletal muscle, central nervous system, intestine and kidney has been suggested from animal studies. Previous experiments in C3 and C4 knockout mice suggested an important role of the classical or lectin pathway in initiation of complement activation during intestinal ischaemia/reperfusion injury while later use of factor D knockout mice showed the alternative pathway to be critically involved. We hypothesized that alternative pathway amplification might play a more critical role in classical pathway-induced C5 activation than previously recognized and used pathway-selective inhibitory mAbs to further elucidate the role of the alternative pathway. Here we demonstrate that selective blockade of the alternative pathway by neutralizing factor D in human serum diluted 1 : 2 with mAb 166-32 inhibited more than 80% of C5a and TCC formation induced by solid phase IgM and solid- and fluid-phase human aggregated IgG via the classical pathway. The findings emphasize the influence of alternative pathway amplification on the effect of initial classical pathway activation and the therapeutic potential of inhibiting the alternative pathway in clinical conditions with excessive and uncontrolled complement activation.
Figures
References
-
- Walport JM. Advances in immunology: Complement – first of two parts. New Engl J Med. 2001;344:1058–66. - PubMed
-
- Mollnes TE, Song W-C, Lambris JD. Complement in inflammatory tissue damage and disease. Trends Immunol. 2002;23:61–4. - PubMed
-
- Weisman HF, Bartow T, Leppo MK, et al. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–51. - PubMed
-
- Chan RK, Ibrahim SI, Verna N, Carroll M, Moore FD, Jr, Hechtman HB. Ischaemia-reperfusion is an event triggered by immune complexes and complement. Br J Surg. 2003;90:1470–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
