

Block of inactivation-deficient Na+ channels by local anesthetics in stably transfected mammalian cells: evidence for drug binding along the activation pathway
- PMID: 15545401
- PMCID: PMC2234030
- DOI: 10.1085/jgp.200409128
Block of inactivation-deficient Na+ channels by local anesthetics in stably transfected mammalian cells: evidence for drug binding along the activation pathway
Abstract
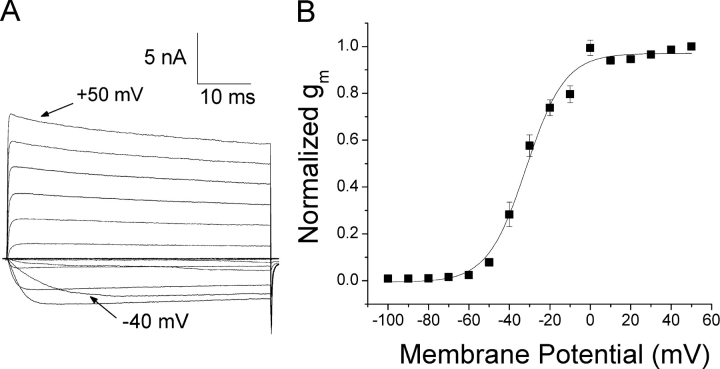
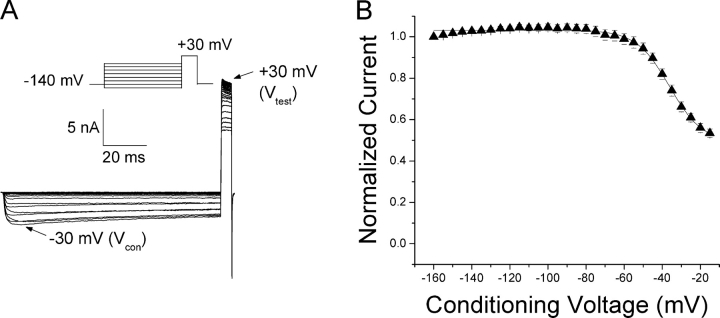
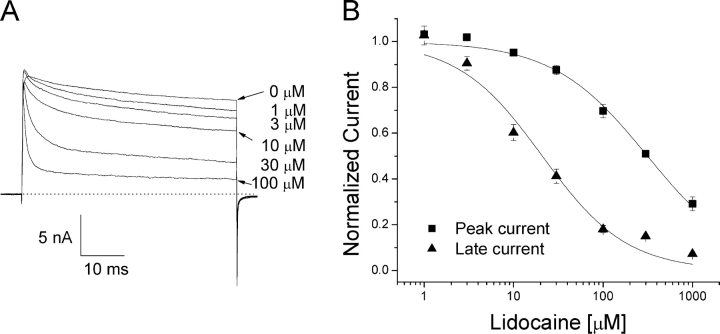
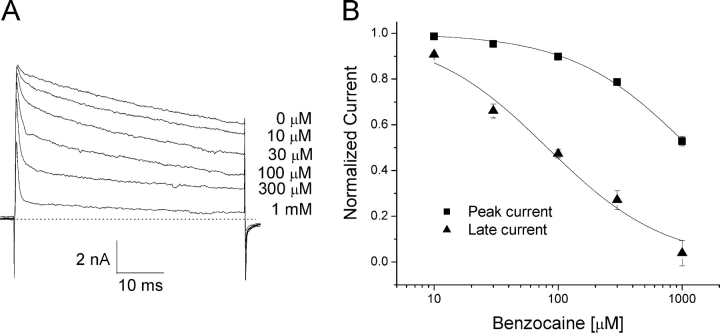
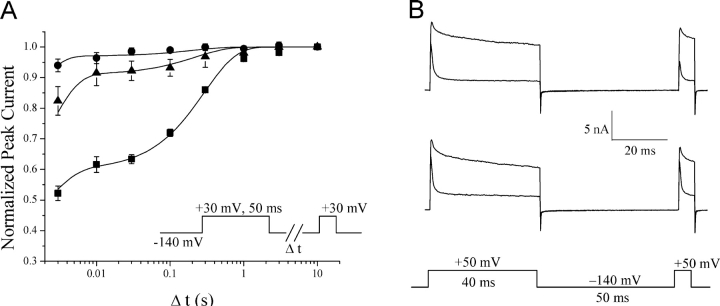
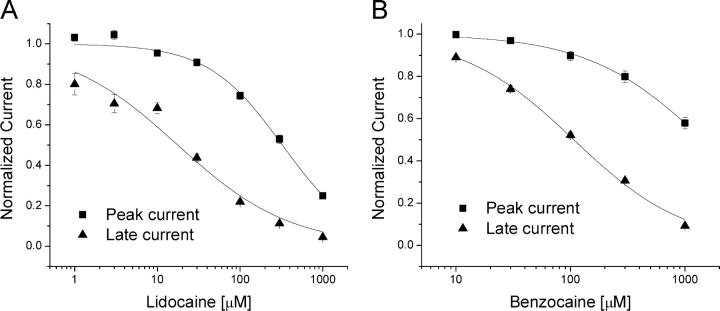
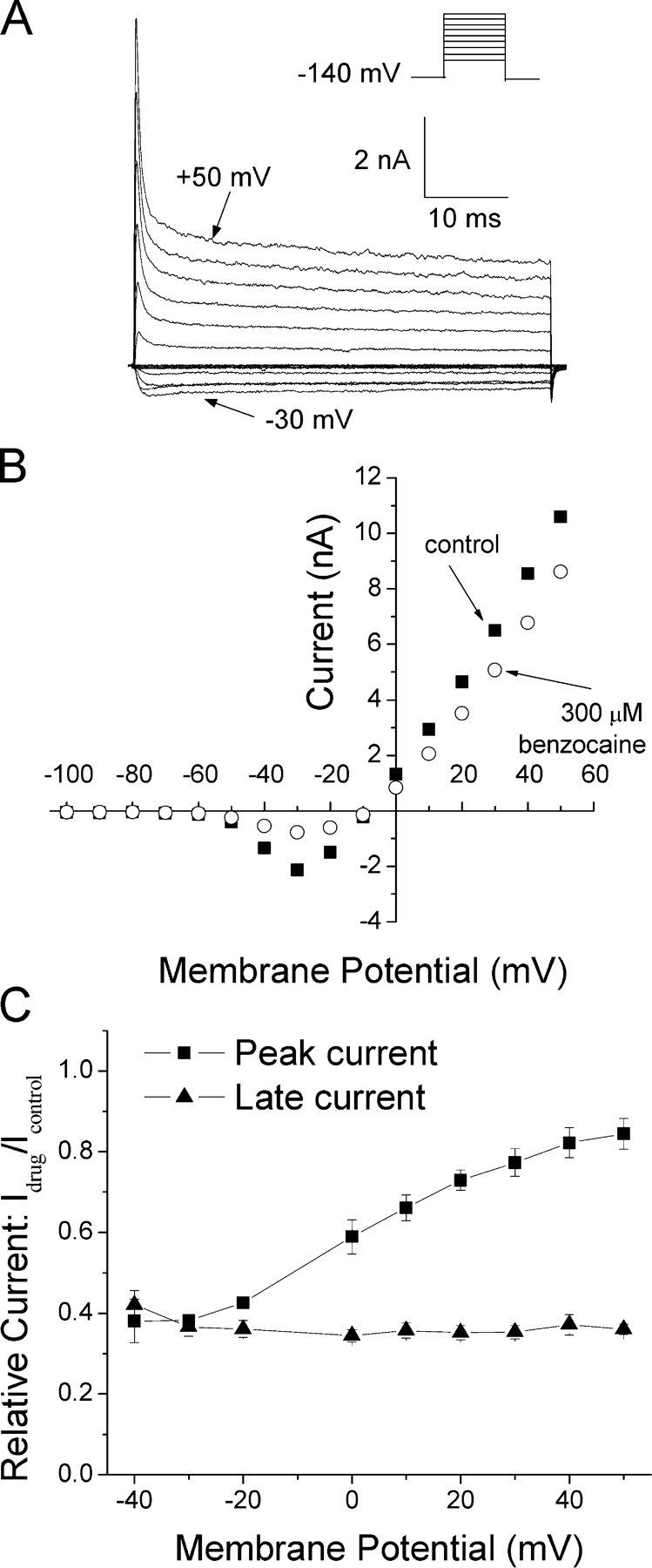
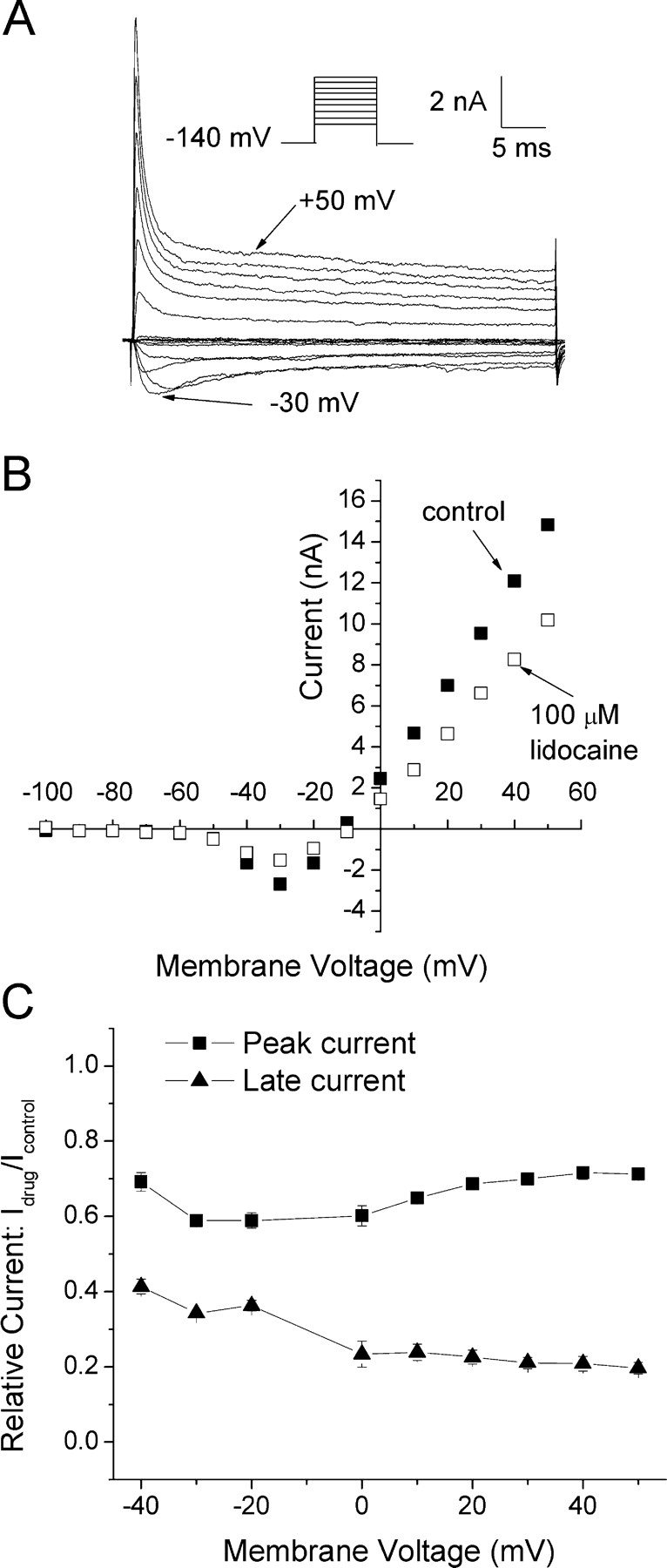
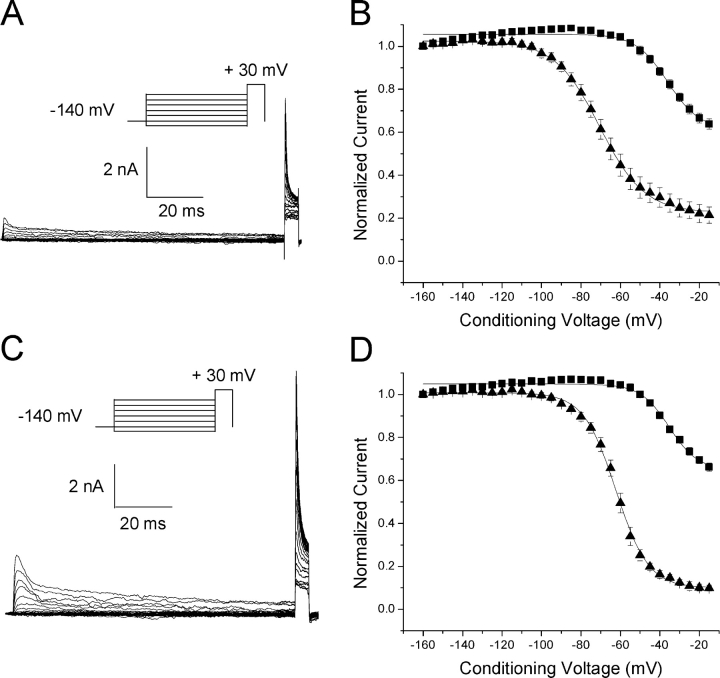
According to the classic modulated receptor hypothesis, local anesthetics (LAs) such as benzocaine and lidocaine bind preferentially to fast-inactivated Na(+) channels with higher affinities. However, an alternative view suggests that activation of Na(+) channels plays a crucial role in promoting high-affinity LA binding and that fast inactivation per se is not a prerequisite for LA preferential binding. We investigated the role of activation in LA action in inactivation-deficient rat muscle Na(+) channels (rNav1.4-L435W/L437C/A438W) expressed in stably transfected Hek293 cells. The 50% inhibitory concentrations (IC(50)) for the open-channel block at +30 mV by lidocaine and benzocaine were 20.9 +/- 3.3 microM (n = 5) and 81.7 +/- 10.6 microM (n = 5), respectively; both were comparable to inactivated-channel affinities. In comparison, IC(50) values for resting-channel block at -140 mV were >12-fold higher than those for open-channel block. With 300 microM benzocaine, rapid time-dependent block (tau approximately 0.8 ms) of inactivation-deficient Na(+) currents occurred at +30 mV, but such a rapid time-dependent block was not evident at -30 mV. The peak current at -30 mV, however, was reduced more severely than that at +30 mV. This phenomenon suggested that the LA block of intermediate closed states took place notably when channel activation was slow. Such closed-channel block also readily accounted for the LA-induced hyperpolarizing shift in the conventional steady-state inactivation measurement. Our data together illustrate that the Na(+) channel activation pathway, including most, if not all, transient intermediate closed states and the final open state, promotes high-affinity LA binding.
Figures
References
-
- Aldrich, R.W., D.P. Corey, and C.F. Stevens. 1983. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 306:436–441. - PubMed
-
- An, R.H., R. Bangalore, S.Z. Rosero, and R.S. Kass. 1996. Lidocaine block of LQT-3 mutant human Na+ channels. Circ. Res. 79:103–108. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
