

A role for proteinase-activated receptor-1 in inflammatory bowel diseases
- PMID: 15545995
- PMCID: PMC526028
- DOI: 10.1172/JCI21689
A role for proteinase-activated receptor-1 in inflammatory bowel diseases
Retraction in
-
A role for proteinase-activated receptor-1 in inflammatory bowel diseases.J Clin Invest. 2006 Jul;116(7):2056. doi: 10.1172/jci21689r1. J Clin Invest. 2006. PMID: 16881139 Free PMC article. No abstract available.
Abstract
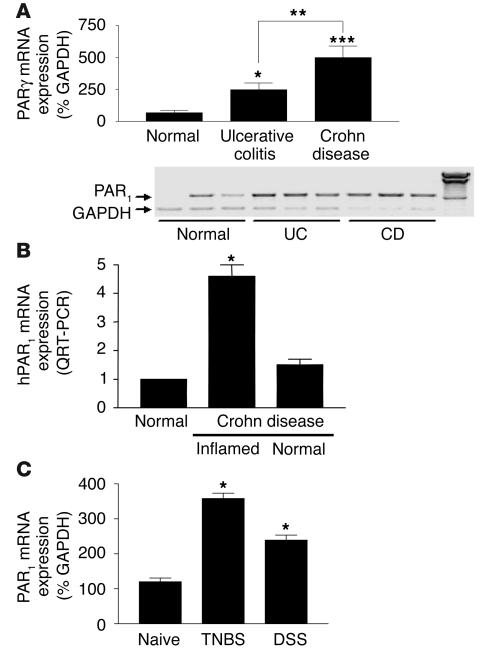
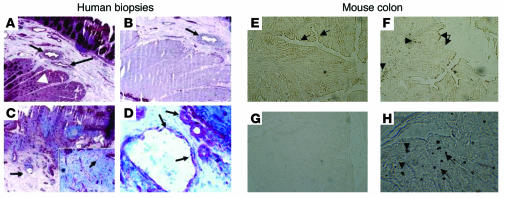
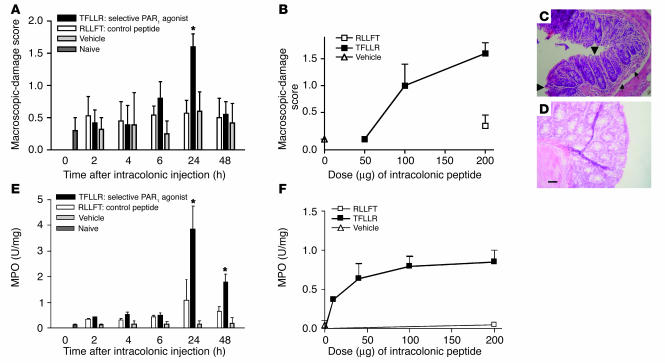
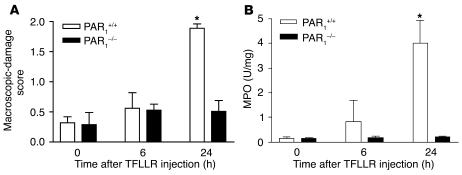
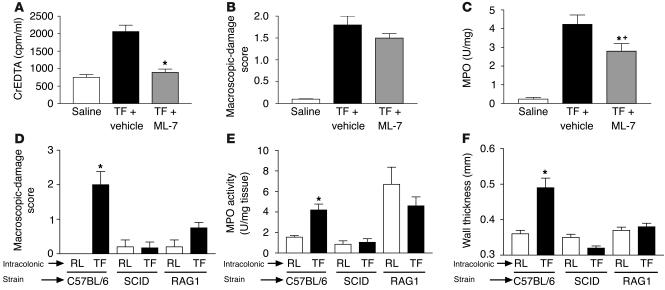
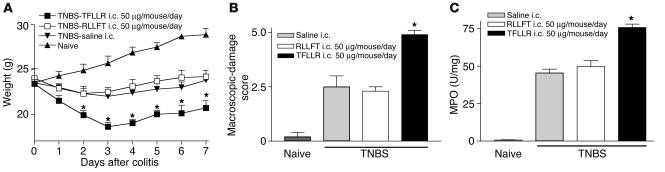
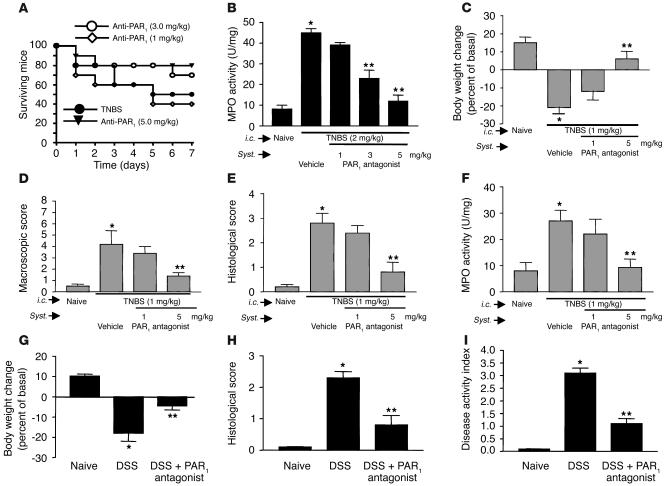
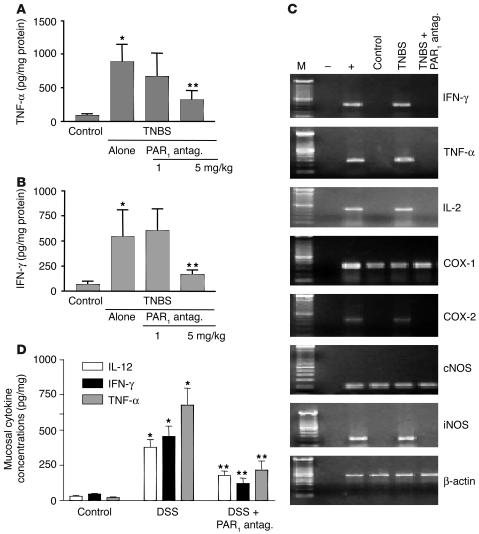
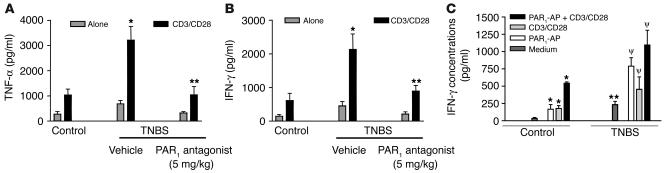
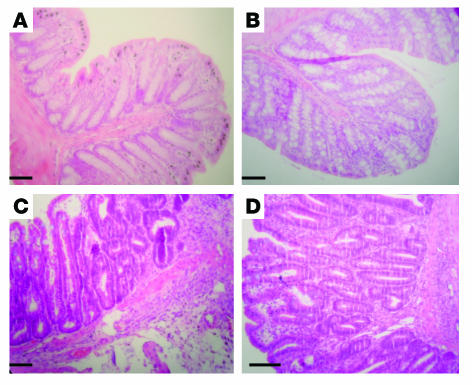
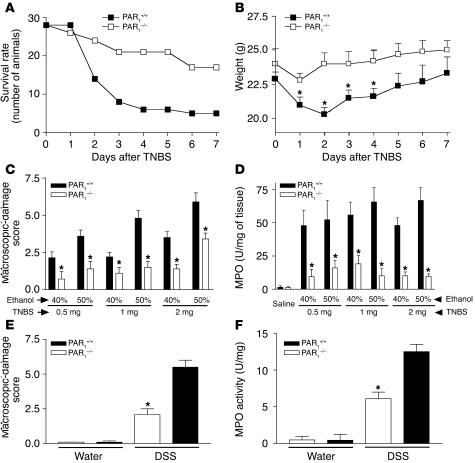
Proteinase-activated receptor-1 (PAR1), a G protein-coupled receptor activated by thrombin, is highly expressed in different cell types of the gastrointestinal tract. The activity of thrombin and of other proteinases is significantly increased in the colon of inflammatory bowel disease (IBD) patients. Since PAR1 activation in tissues other than the gut provoked inflammation, we hypothesized that PAR1 activation in the colon is involved in the pathogenesis of IBD. Here, we demonstrate that PAR1 is overexpressed in the colon of IBD patients. In mice, intracolonic administration of PAR1 agonists led to an inflammatory reaction characterized by edema and granulocyte infiltration. This PAR1 activation-induced inflammation was dependent on B and T lymphocytes. Moreover, PAR1 activation exacerbated and prolonged inflammation in a mouse model of IBD induced by the intracolonic administration of trinitrobenzene sulfonic acid (TNBS), while PAR1 antagonism significantly decreased the mortality and severity of colonic inflammation induced by TNBS and dextran sodium sulfate. In these 2 models, colitis development was strongly attenuated by PAR1 deficiency. Taken together, these results imply an important role for PAR1 in the pathogenesis of experimental colitis, supporting the notion that PAR1 inhibition may be beneficial in the context of IBD and possibly in other chronic intestinal inflammatory disorders.
Figures
Similar articles
-
Protease-activated receptor-2 activation: a major actor in intestinal inflammation.Gut. 2008 Sep;57(9):1222-9. doi: 10.1136/gut.2008.150722. Epub 2008 May 6. Gut. 2008. PMID: 18460552
-
R-spondin1, a novel intestinotrophic mitogen, ameliorates experimental colitis in mice.Gastroenterology. 2007 Apr;132(4):1331-43. doi: 10.1053/j.gastro.2007.02.001. Epub 2007 Feb 6. Gastroenterology. 2007. PMID: 17408649
-
Curcumin prevents and ameliorates trinitrobenzene sulfonic acid-induced colitis in mice.Gastroenterology. 2002 Dec;123(6):1912-22. doi: 10.1053/gast.2002.37050. Gastroenterology. 2002. PMID: 12454848
-
Experimental models of inflammatory bowel disease.Arch Immunol Ther Exp (Warsz). 2003;51(3):149-55. Arch Immunol Ther Exp (Warsz). 2003. PMID: 12894869 Review.
-
Role of microecology in chronic inflammatory bowel diseases.Eur J Clin Nutr. 2002 Dec;56 Suppl 4:S34-8. doi: 10.1038/sj.ejcn.1601662. Eur J Clin Nutr. 2002. PMID: 12556945 Review.
Cited by
-
The thrombin receptor links brain derived neurotrophic factor to neuron cholesterol production, resiliency and repair after spinal cord injury.Neurobiol Dis. 2021 May;152:105294. doi: 10.1016/j.nbd.2021.105294. Epub 2021 Feb 5. Neurobiol Dis. 2021. PMID: 33549720 Free PMC article.
-
Protease Activated Receptor 4 as a Novel Modulator of Regulatory T Cell Function.Front Immunol. 2019 Jun 18;10:1311. doi: 10.3389/fimmu.2019.01311. eCollection 2019. Front Immunol. 2019. PMID: 31275306 Free PMC article.
-
Functional GI disorders: from animal models to drug development.Gut. 2008 Mar;57(3):384-404. doi: 10.1136/gut.2006.101675. Epub 2007 Oct 26. Gut. 2008. PMID: 17965064 Free PMC article. Review.
-
Insulin-like growth factor-1 receptor transactivation modulates the inflammatory and proliferative responses of neurotensin in human colonic epithelial cells.J Biol Chem. 2011 Feb 25;286(8):6092-9. doi: 10.1074/jbc.M110.192534. Epub 2011 Jan 6. J Biol Chem. 2011. PMID: 21212273 Free PMC article.
-
Clinical relevance of proteinase activated receptors (pars) in the gut.Gut. 2005 Jun;54(6):867-74. doi: 10.1136/gut.2004.048876. Gut. 2005. PMID: 15888798 Free PMC article. Review. No abstract available.
References
-
- Hollenberg MD, Compton SJ. International Union of Pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol. Rev. 2002;54:203–217. - PubMed
-
- Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol. Rev. 2001;53:245–282. - PubMed
-
- Hollenberg MD, Saifeddine M, Al-Ani B, Kawabata A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can. J. Physiol. Pharmacol. 1997;75:832–841. - PubMed
-
- Vergnolle N, Wallace JL, Bunnett NW, Hollenberg MD. Protease-activated receptors in inflammation, neuronal signaling and pain. Trends Pharmacol. Sci. 2001;22:146–152. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
