

On the mechanism and biology of cytochrome oxidase inhibition by nitric oxide
- PMID: 15546991
- PMCID: PMC534717
- DOI: 10.1073/pnas.0405368101
On the mechanism and biology of cytochrome oxidase inhibition by nitric oxide
Abstract
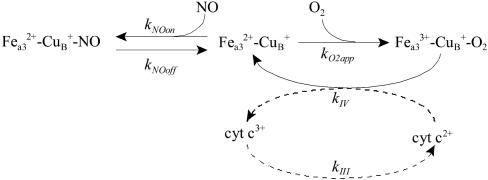
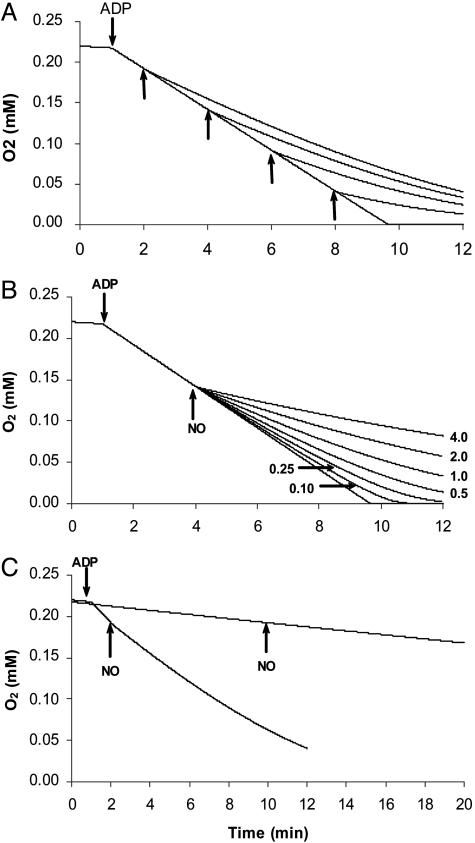
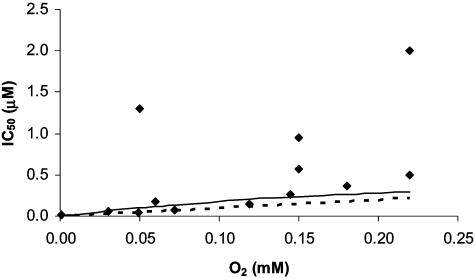
The detailed molecular mechanism for the reversible inhibition of mitochondrial respiration by NO has puzzled investigators: The rate constants for the binding of NO and O2 to the reduced binuclear center CuB/a3 of cytochrome oxidase (COX) are similar, and NO is able to dissociate slowly from this center whereas O2 is kinetically trapped, which altogether seems to favor the complex of COX with O2 over the complex of COX with NO. Paradoxically, the inhibition of COX by NO is observed at high ratios of O2 to NO (in the 40-500 range) and is very fast (seconds or faster). In this work, we used simple mathematical models to investigate this paradox and other important biological questions concerning the inhibition of COX by NO. The results showed that all known features of the inhibition of COX by NO can be accounted for by a direct competition between NO and O2 for the reduced binuclear center CuB/a3 of COX. Besides conciliating apparently contradictory data, this work provided an explanation for the so-called excess capacity of COX by showing that the COX activity found in tissues actually is optimized to avoid an excessive inhibition of mitochondrial respiration by NO, allowing a moderate, but not excessive, overlap between the roles of NO in COX inhibition and in cellular signaling. In pathological situations such as COX-deficiency diseases and chronic inflammation, an excessive inhibition of the mitochondrial respiration is predicted.
Figures


References
-
- Brown, G. C. & Cooper, C. E. (1994) FEBS Lett. 356, 295–298. - PubMed
-
- Cleeter, M. J. W., Cooper, J. M., Darley-Usmar, V. M., Moncada, S. & Schapira, A. H. V. (1994) FEBS Lett. 345, 50–54. - PubMed
-
- Poderoso, J. J., Carreras, M. C., Lisdero, C., Riobo, N., Schopfer, F. & Boveris, A. (1996) Arch. Biochem. Biophys. 328, 85–92. - PubMed
-
- Boveris, A., Costa, L. E., Poderoso, J. J., Carreras, M. C. & Cadenas, E. (2000) Ann. N.Y. Acad. Sci. 899, 121–135. - PubMed
-
- Moncada, S. & Erusalimsky, J. D. (2002) Nat. Rev. Mol. Cell Biol. 3, 214–220. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
