

GOtcha: a new method for prediction of protein function assessed by the annotation of seven genomes
- PMID: 15550167
- PMCID: PMC535938
- DOI: 10.1186/1471-2105-5-178
GOtcha: a new method for prediction of protein function assessed by the annotation of seven genomes
Abstract
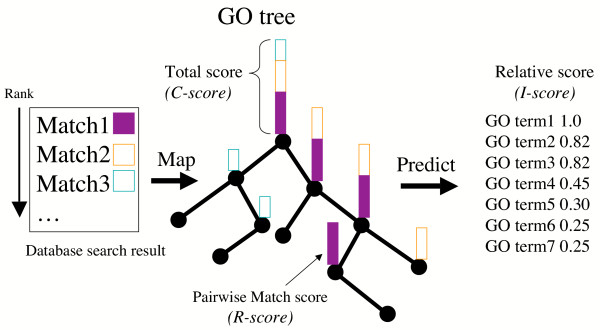
Background: The function of a novel gene product is typically predicted by transitive assignment of annotation from similar sequences. We describe a novel method, GOtcha, for predicting gene product function by annotation with Gene Ontology (GO) terms. GOtcha predicts GO term associations with term-specific probability (P-score) measures of confidence. Term-specific probabilities are a novel feature of GOtcha and allow the identification of conflicts or uncertainty in annotation.
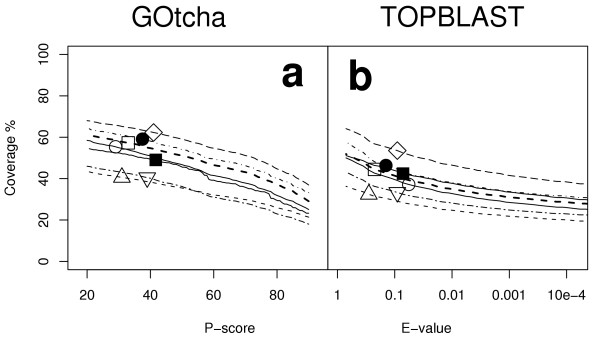
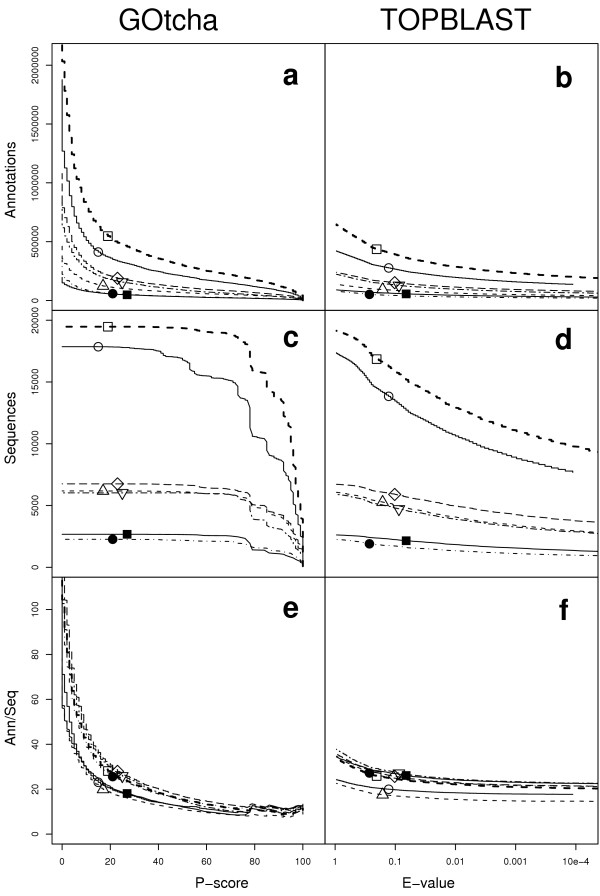
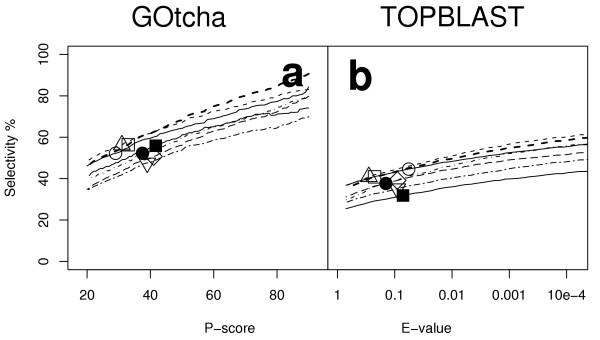
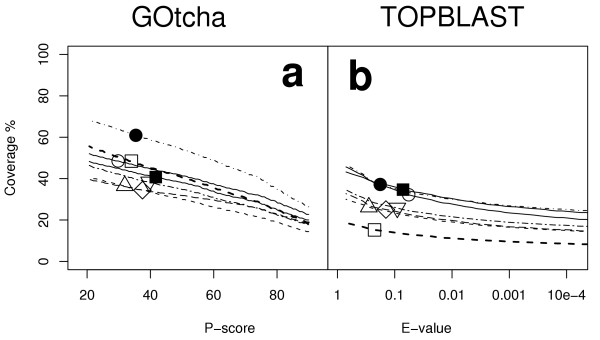
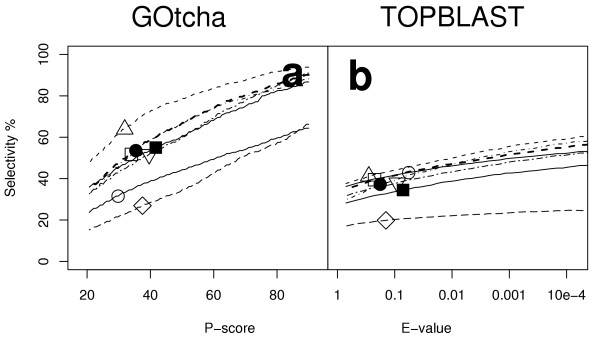
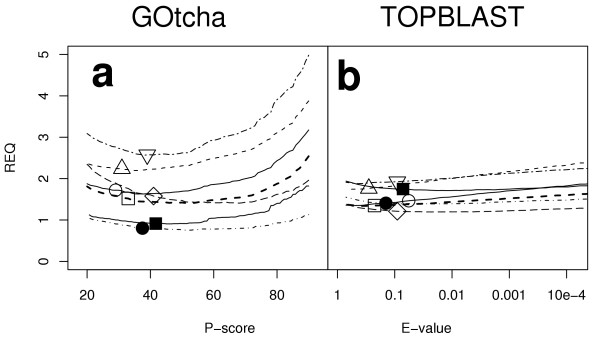
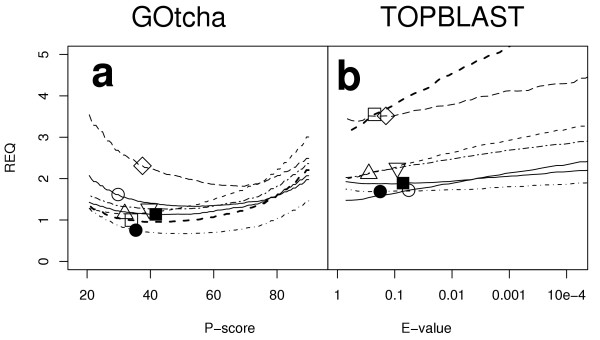
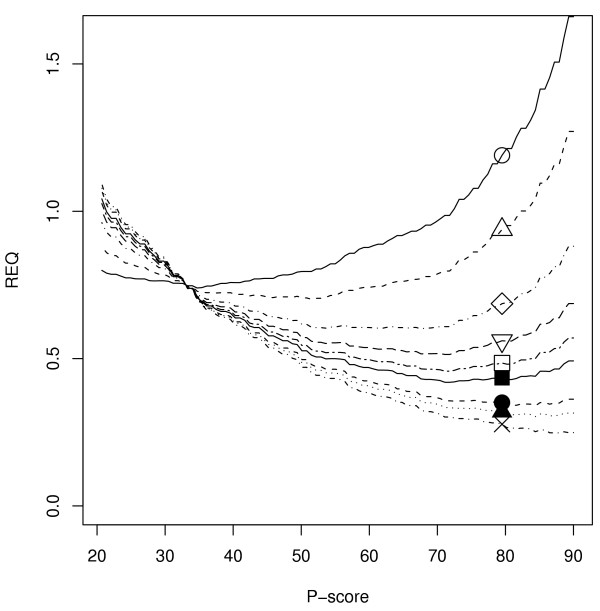
Results: The GOtcha method was applied to the recently sequenced genome for Plasmodium falciparum and six other genomes. GOtcha was compared quantitatively for retrieval of assigned GO terms against direct transitive assignment from the highest scoring annotated BLAST search hit (TOPBLAST). GOtcha exploits information deep into the 'twilight zone' of similarity search matches, making use of much information that is otherwise discarded by more simplistic approaches. At a P-score cutoff of 50%, GOtcha provided 60% better recovery of annotation terms and 20% higher selectivity than annotation with TOPBLAST at an E-value cutoff of 10(-4).
Conclusions: The GOtcha method is a useful tool for genome annotators. It has identified both errors and omissions in the original Plasmodium falciparum annotation and is being adopted by many other genome sequencing projects.
Figures
References
-
- Ashburner M, Ball C, Blake J, Botstein D, Butler H, Cherry J, Davis A, Dolinski K, Dwight S, Eppig J, Harris M, Hill D, Issel-Tarver L, Kasarkis A, Lewis S, Matese J, Richardson J, Ringwald M, Rubin G, Sherlock G. Gene Ontology: Tool for the Unification of Biology. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
