

Furin-mediated cleavage of the feline foamy virus Env leader protein
- PMID: 15564468
- PMCID: PMC533928
- DOI: 10.1128/JVI.78.24.13573-13581.2004
Furin-mediated cleavage of the feline foamy virus Env leader protein
Abstract
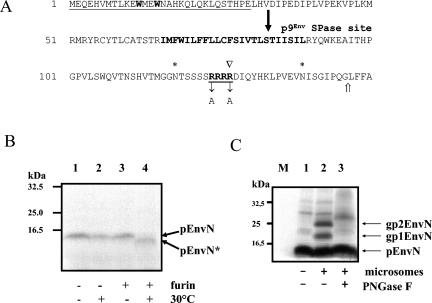
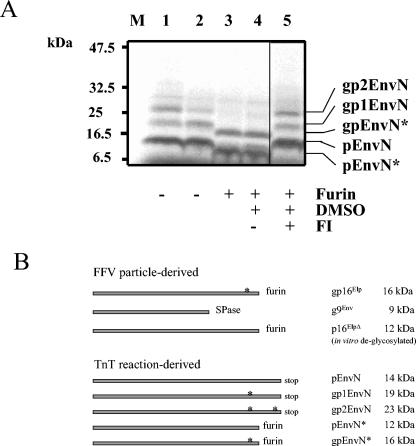
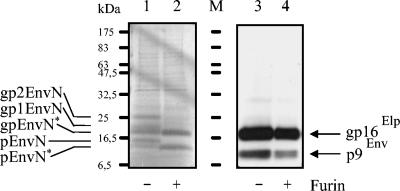
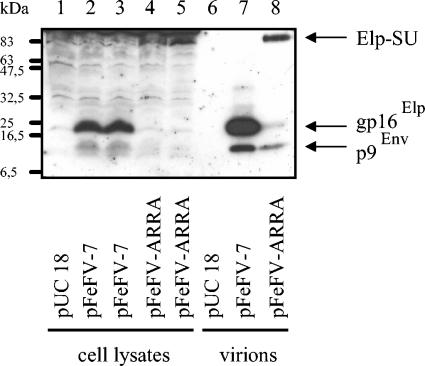
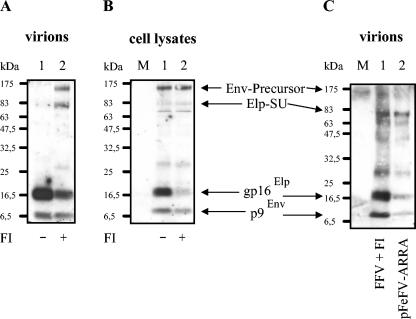
The molecular biology of spuma or foamy retroviruses is different from that of the other members of the Retroviridae. Among the distinguishing features, the N-terminal domain of the foamy virus Env glycoprotein, the 16-kDa Env leader protein Elp, is a component of released, infectious virions and is required for particle budding. The transmembrane protein Elp specifically interacts with N-terminal Gag sequences during morphogenesis. In this study, we investigate the mechanism of Elp release from the Env precursor protein. By a combination of genetic, biochemical, and biophysical methods, we show that the feline foamy virus (FFV) Elp is released by a cellular furin-like protease, most likely furin itself, generating an Elp protein consisting of 127 amino acid residues. The cleavage site fully conforms to the rules for an optimal furin site. Proteolytic processing at the furin cleavage site is required for full infectivity of FFV. However, utilization of other furin proteases and/or cleavage at a suboptimal signal peptidase cleavage site can partially rescue virus viability. In addition, we show that FFV Elp carries an N-linked oligosaccharide that is not conserved among the known foamy viruses.
Figures


Similar articles
-
Prototype foamy virus envelope glycoprotein leader peptide processing is mediated by a furin-like cellular protease, but cleavage is not essential for viral infectivity.J Virol. 2004 Dec;78(24):13865-70. doi: 10.1128/JVI.78.24.13865-13870.2004. J Virol. 2004. PMID: 15564494 Free PMC article.
-
Features of the Env leader protein and the N-terminal Gag domain of feline foamy virus important for virus morphogenesis.Virology. 2003 Jun 5;310(2):235-44. doi: 10.1016/s0042-6822(03)00125-9. Virology. 2003. PMID: 12781711
-
Mutagenesis of N-terminal residues of feline foamy virus Gag reveals entirely distinct functions during capsid formation, particle assembly, Gag processing and budding.Retrovirology. 2016 Aug 22;13(1):57. doi: 10.1186/s12977-016-0291-8. Retrovirology. 2016. PMID: 27549192 Free PMC article.
-
The foamy virus envelope glycoproteins.Curr Top Microbiol Immunol. 2003;277:111-29. doi: 10.1007/978-3-642-55701-9_5. Curr Top Microbiol Immunol. 2003. PMID: 12908770 Review.
-
Proteolytic processing of foamy virus Gag and Pol proteins.Curr Top Microbiol Immunol. 2003;277:63-88. doi: 10.1007/978-3-642-55701-9_3. Curr Top Microbiol Immunol. 2003. PMID: 12908768 Review.
Cited by
-
Ubiquitination of the prototype foamy virus envelope glycoprotein leader peptide regulates subviral particle release.J Virol. 2005 Dec;79(24):15074-83. doi: 10.1128/JVI.79.24.15074-15083.2005. J Virol. 2005. PMID: 16306578 Free PMC article.
-
The Bombyx mori Nucleopolyhedrovirus GP64 Retains the Transmembrane Helix of Signal Peptide to Contribute to Secretion across the Cytomembrane.Microbiol Spectr. 2022 Aug 31;10(4):e0191322. doi: 10.1128/spectrum.01913-22. Epub 2022 Aug 8. Microbiol Spectr. 2022. PMID: 35938817 Free PMC article.
-
Early events in foamy virus-host interaction and intracellular trafficking.Viruses. 2013 Apr 8;5(4):1055-74. doi: 10.3390/v5041055. Viruses. 2013. PMID: 23567621 Free PMC article. Review.
-
Foamy virus budding and release.Viruses. 2013 Apr 10;5(4):1075-98. doi: 10.3390/v5041075. Viruses. 2013. PMID: 23575110 Free PMC article. Review.
-
Non-simian foamy viruses: molecular virology, tropism and prevalence and zoonotic/interspecies transmission.Viruses. 2013 Sep 13;5(9):2169-209. doi: 10.3390/v5092169. Viruses. 2013. PMID: 24064793 Free PMC article. Review.
References
-
- Alke, A., A. Schwantes, K. Kido, M. Flötenmeyer, R. M. Flügel, and M. Löchelt. 2001. The bet gene of feline foamy virus is required for virus replication. Virology 287:310-320. - PubMed
-
- Alke, A., A. Schwantes, M. Zemba, R. M. Flügel, and M. Löchelt. 2000. Characterization of the humoral immune response and virus replication in cats experimentally infected with feline foamy virus. Virology 275:170-176. - PubMed
-
- Basak, A., M. Zhong, J. Munzer, M. Chretien, and N. Seidah. 2001. Implication of the proprotein convertases furin, PC5 and PC7 in the cleavage of surface glycoproteins of Hong Kong, Ebola and respiratory syncytial viruses: a comparative analysis with fluorogenic peptides. Biochem. J. 353:537-545. - PMC - PubMed
-
- Bornsen, K. 2000. Influence of salts, buffers, detergents, solvents, and matrices on MALDI-MS protein analysis in complex mixtures. Methods Mol. Biol. 146:387-404. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
