

Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation
- PMID: 15564577
- PMCID: PMC2945223
- DOI: 10.1523/JNEUROSCI.2469-04.2004
Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation
Abstract
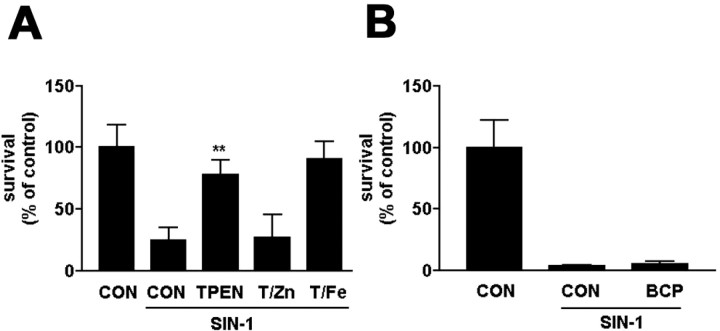
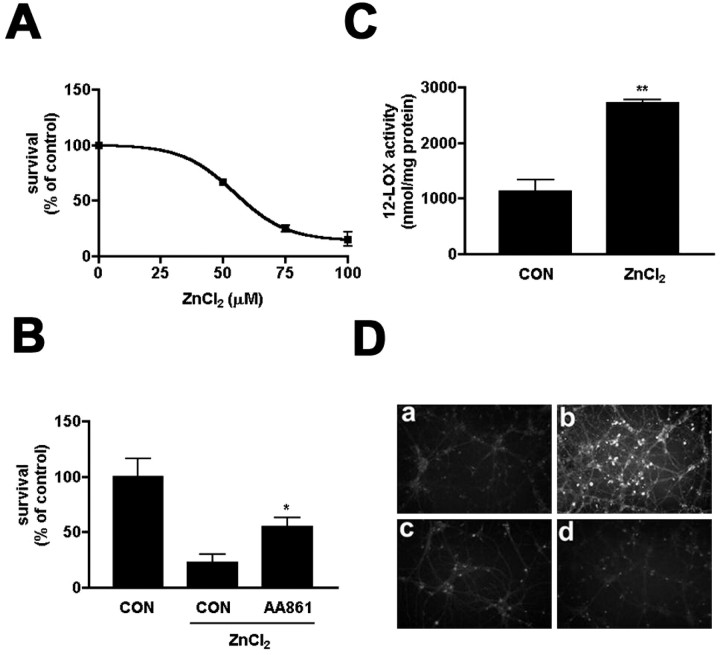
Peroxynitrite toxicity is a major cause of neuronal injury in stroke and neurodegenerative disorders. The mechanisms underlying the neurotoxicity induced by peroxynitrite are still unclear. In this study, we observed that TPEN [N,N,N',N'-tetrakis (2-pyridylmethyl)ethylenediamine], a zinc chelator, protected against neurotoxicity induced by exogenous as well as endogenous (coadministration of NMDA and a nitric oxide donor, diethylenetriamine NONOate) peroxynitrite. Two different approaches to detecting intracellular zinc release demonstrated the liberation of zinc from intracellular stores by peroxynitrite. In addition, we found that peroxynitrite toxicity was blocked by inhibitors of 12-lipoxygenase (12-LOX), p38 mitogen-activated protein kinase (MAPK), and caspase-3 and was associated with mitochondrial membrane depolarization. Inhibition of 12-LOX blocked the activation of p38 MAPK and caspase-3. Zinc itself induced the activation of 12-LOX, generation of reactive oxygen species (ROS), and activation of p38 MAPK and caspase-3. These data suggest a cell death pathway triggered by peroxynitrite in which intracellular zinc release leads to activation of 12-LOX, ROS accumulation, p38 activation, and caspase-3 activation. Therefore, therapies aimed at maintaining intracellular zinc homeostasis or blocking activation of 12-LOX may provide a novel avenue for the treatment of inflammation, stroke, and neurodegenerative diseases in which the formation of peroxynitrite is thought to be one of the important causes of cell death.
Figures








Similar articles
-
Intracellular zinc release and ERK phosphorylation are required upstream of 12-lipoxygenase activation in peroxynitrite toxicity to mature rat oligodendrocytes.J Biol Chem. 2006 Apr 7;281(14):9460-70. doi: 10.1074/jbc.M510650200. Epub 2006 Jan 23. J Biol Chem. 2006. PMID: 16431921
-
Inhibition of poly(ADP-ribose) synthetase (PARS) and protection against peroxynitrite-induced cytotoxicity by zinc chelation.Br J Pharmacol. 1999 Feb;126(3):769-77. doi: 10.1038/sj.bjp.0702332. Br J Pharmacol. 1999. PMID: 10188990 Free PMC article.
-
The involvement of caspase-11 in TPEN-induced apoptosis.FEBS Lett. 2008 Jun 11;582(13):1871-6. doi: 10.1016/j.febslet.2008.04.056. Epub 2008 May 12. FEBS Lett. 2008. PMID: 18474237
-
Intracellular zinc release, 12-lipoxygenase activation and MAPK dependent neuronal and oligodendroglial death.Mol Med. 2007 Jul-Aug;13(7-8):350-5. doi: 10.2119/2007–00042.Zhang. Mol Med. 2007. PMID: 17622306 Free PMC article. Review.
-
Nitrosative stress and potassium channel-mediated neuronal apoptosis: is zinc the link?Pflugers Arch. 2004 Jun;448(3):296-303. doi: 10.1007/s00424-004-1256-7. Epub 2004 Mar 13. Pflugers Arch. 2004. PMID: 15024658 Free PMC article. Review.
Cited by
-
The ZIP3 Zinc Transporter Is Localized to Mossy Fiber Terminals and Is Required for Kainate-Induced Degeneration of CA3 Neurons.J Neurosci. 2022 Mar 30;42(13):2824-2834. doi: 10.1523/JNEUROSCI.0908-21.2022. Epub 2022 Feb 15. J Neurosci. 2022. PMID: 35169020 Free PMC article.
-
Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies.Prog Lipid Res. 2019 Jan;73:28-45. doi: 10.1016/j.plipres.2018.11.001. Epub 2018 Nov 22. Prog Lipid Res. 2019. PMID: 30472260 Free PMC article. Review.
-
Cancer Microenvironment and Endoplasmic Reticulum Stress Response.Mediators Inflamm. 2015;2015:417281. doi: 10.1155/2015/417281. Epub 2015 Sep 27. Mediators Inflamm. 2015. PMID: 26491226 Free PMC article. Review.
-
Glutamate gradually elevates [Zn2+]i via the CaM-CaMKII-NOS cascade in primary cultured rat embryonic cortical neurons.Sci Rep. 2025 Apr 30;15(1):15205. doi: 10.1038/s41598-025-99142-1. Sci Rep. 2025. PMID: 40307298 Free PMC article.
-
Pathophysiology of glia in perinatal white matter injury.Glia. 2014 Nov;62(11):1790-815. doi: 10.1002/glia.22658. Epub 2014 Mar 31. Glia. 2014. PMID: 24687630 Free PMC article. Review.
References
-
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ (2000) Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem 75: 1878-1888. - PubMed
-
- Arai K, Nishiyama N, Matsuki N, Ikegaya Y (2001) Neuroprotective effects of lipoxygenase inhibitors against ischemic injury in rat hippocampal slice cultures. Brain Res 904: 167-172. - PubMed
-
- Armstrong C, Leong W, Lees GJ (2001) Comparative effects of metal chelating agents on the neuronal cytotoxicity induced by copper (Cu+2), iron (Fe+3) and zinc in the hippocampus. Brain Res 892: 51-62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials