

Structure and evolution of protein interaction networks: a statistical model for link dynamics and gene duplications
- PMID: 15566577
- PMCID: PMC544576
- DOI: 10.1186/1471-2148-4-51
Structure and evolution of protein interaction networks: a statistical model for link dynamics and gene duplications
Abstract
Background: The structure of molecular networks derives from dynamical processes on evolutionary time scales. For protein interaction networks, global statistical features of their structure can now be inferred consistently from several large-throughput datasets. Understanding the underlying evolutionary dynamics is crucial for discerning random parts of the network from biologically important properties shaped by natural selection.
Results: We present a detailed statistical analysis of the protein interactions in Saccharomyces cerevisiae based on several large-throughput datasets. Protein pairs resulting from gene duplications are used as tracers into the evolutionary past of the network. From this analysis, we infer rate estimates for two key evolutionary processes shaping the network: (i) gene duplications and (ii) gain and loss of interactions through mutations in existing proteins, which are referred to as link dynamics. Importantly, the link dynamics is asymmetric, i.e., the evolutionary steps are mutations in just one of the binding parters. The link turnover is shown to be much faster than gene duplications. Both processes are assembled into an empirically grounded, quantitative model for the evolution of protein interaction networks.
Conclusions: According to this model, the link dynamics is the dominant evolutionary force shaping the statistical structure of the network, while the slower gene duplication dynamics mainly affects its size. Specifically, the model predicts (i) a broad distribution of the connectivities (i.e., the number of binding partners of a protein) and (ii) correlations between the connectivities of interacting proteins, a specific consequence of the asymmetry of the link dynamics. Both features have been observed in the protein interaction network of S. cerevisiae.
Figures

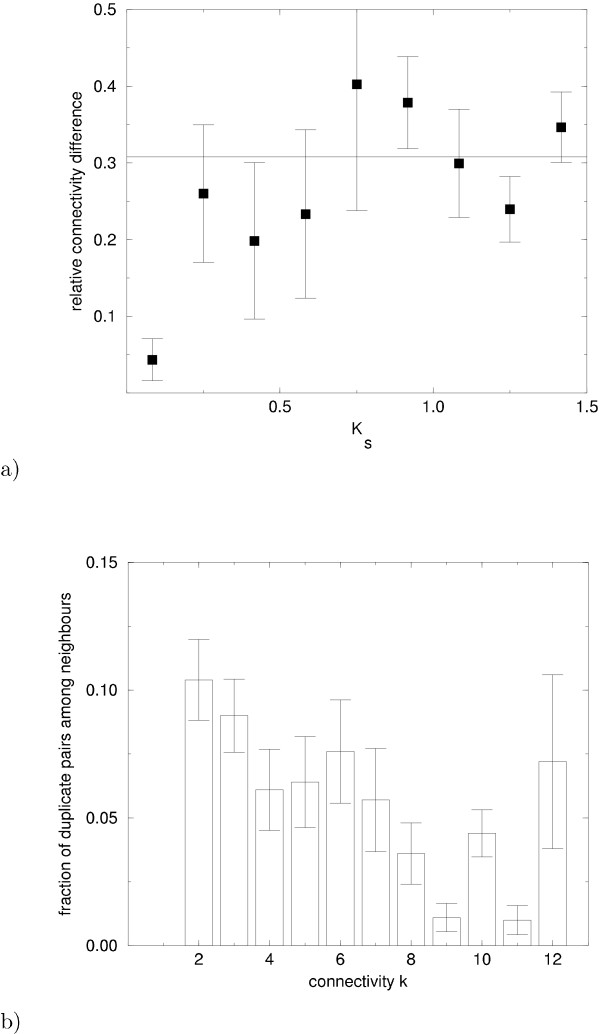

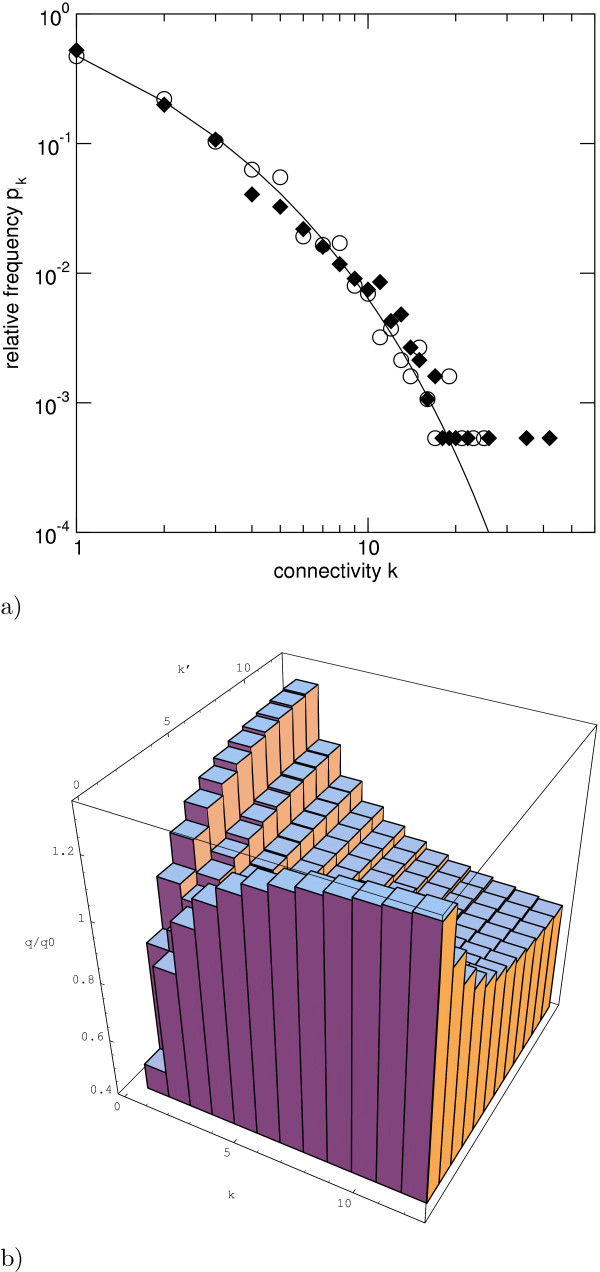
 and the 'null distribution'
and the 'null distribution'  of an uncorrelated random network, see text.
of an uncorrelated random network, see text.References
-
- Albert R, Barabási AL. Statistical mechanics of complex networks. Rev Mod Phys. 2002;74:47–97. doi: 10.1103/RevModPhys.74.47. - DOI
-
- Dorogovtsev SN, Mendes JFF. Evolution of Networks. Adv Phys. 2002;51:1079–1187. doi: 10.1080/00018730110112519. - DOI
-
- Newman MEJ. The structure and function of complex networks. SIAM Review. 2003;45:167–256.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
