

Computing prokaryotic gene ubiquity: rescuing the core from extinction
- PMID: 15574825
- PMCID: PMC534671
- DOI: 10.1101/gr.3024704
Computing prokaryotic gene ubiquity: rescuing the core from extinction
Abstract
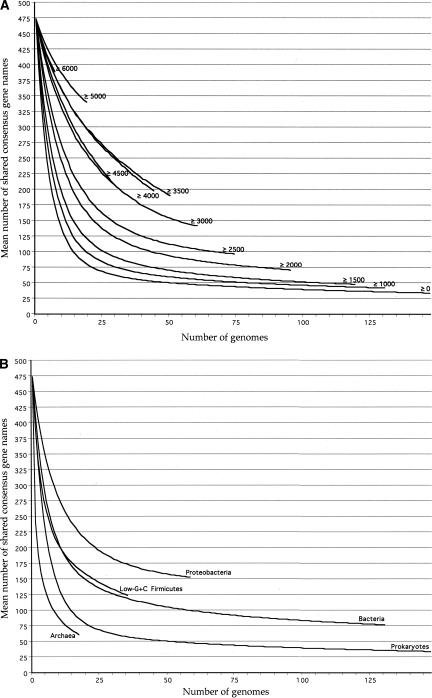
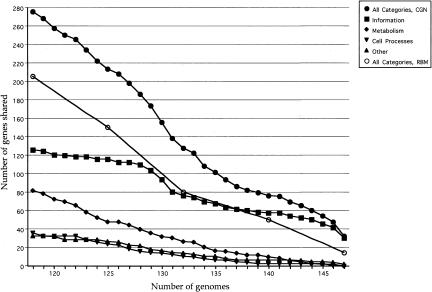
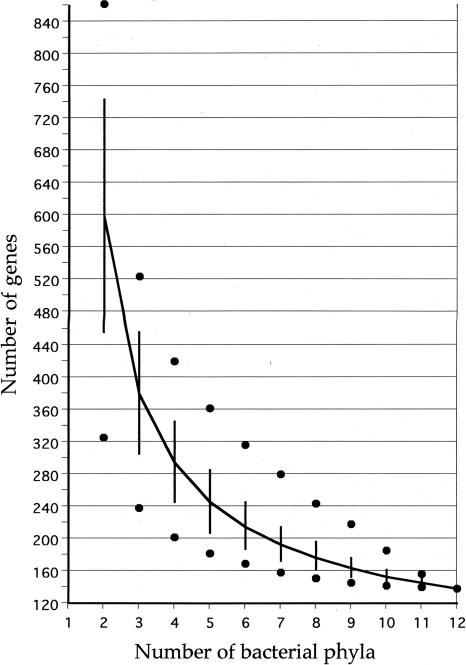
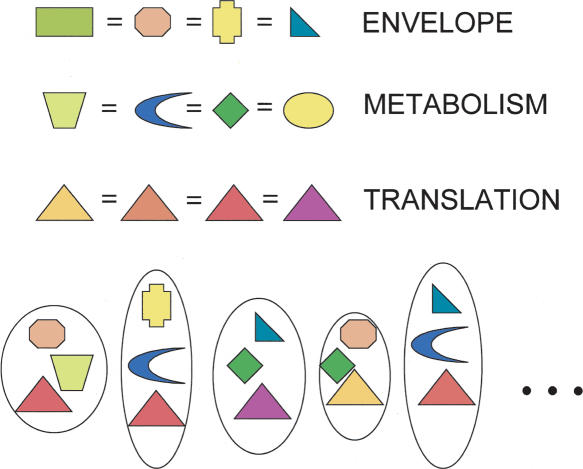
The genomic core concept has found several uses in comparative and evolutionary genomics. Defined as the set of all genes common to (ubiquitous among) all genomes in a phylogenetically coherent group, core size decreases as the number and phylogenetic diversity of the relevant group increases. Here, we focus on methods for defining the size and composition of the core of all genes shared by sequenced genomes of prokaryotes (Bacteria and Archaea). There are few (almost certainly less than 50) genes shared by all of the 147 genomes compared, surely insufficient to conduct all essential functions. Sequencing and annotation errors are responsible for the apparent absence of some genes, while very limited but genuine disappearances (from just one or a few genomes) can account for several others. Core size will continue to decrease as more genome sequences appear, unless the requirement for ubiquity is relaxed. Such relaxation seems consistent with any reasonable biological purpose for seeking a core, but it renders the problem of definition more problematic. We propose an alternative approach (the phylogenetically balanced core), which preserves some of the biological utility of the core concept. Cores, however delimited, preferentially contain informational rather than operational genes; we present a new hypothesis for why this might be so.
Figures
References
-
- Boucher, Y., Douady, C.J., Papke, R.T., Walsh, D.A., Boudreau, M.E., Nesbø, C.L., Case, R.J., and Doolittle, W.F. 2003. Lateral gene transfer and the origins of prokaryotic groups. Annu. Rev. Genet. 37: 283-328. - PubMed
-
- Brochier, C., Bapteste, E., Moreira, D., and Philippe, H. 2002. Eubacterial phylogeny based on translational apparatus proteins. Trends Genet. 18: 1-5. - PubMed
-
- Brown, J.R., Douady, C.J., Italia, M.J., Marshall, W.E., and Stanhope, M.J. 2001. Universal trees based on large combined protein sequence data sets. Nat. Genet. 28: 281-285. - PubMed
-
- Charlebois, R.L., Clarke, G.D.P., Beiko, R.G., and St. Jean, A. 2003. Characterization of species-specific genes using a flexible, web-based querying system. FEMS Microbiol. Lett. 225: 213-220. - PubMed
Web site references
-
- http://www.neurogadgets.com/bws.php; The NeuroGadgets Inc. Bioinformatics Web Service (NGIBWS).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical