

Computational analysis of the transient movement of helices in sensory rhodopsin II
- PMID: 15576566
- PMCID: PMC2253333
- DOI: 10.1110/ps.04973805
Computational analysis of the transient movement of helices in sensory rhodopsin II
Abstract
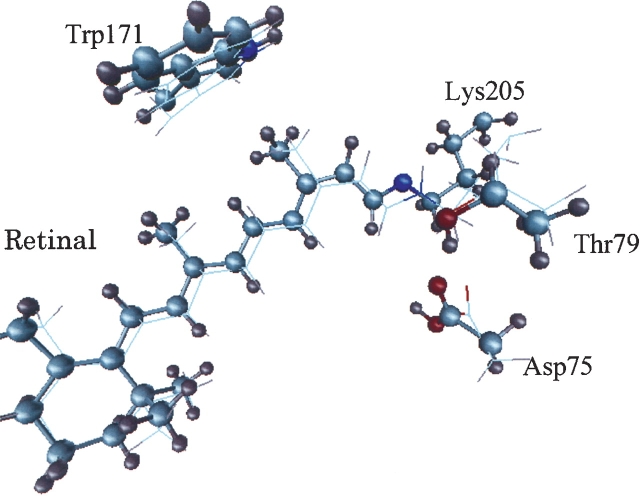
MD simulation of sensory rhodopsin II was executed for three intermediates (ground-state, K-state, M-state) appearing in its photocycle. We observed a large displacement of the cytoplasmic side of helixF only in M-state among the three intermediates. This displacement was transmitted to TM2, and the cytoplasmic side of TM2 rotated clockwise. These transient movements are in agreement with the results of an EPR experiment. That is, the early stage of signal transduction in a sRII-HtrII complex was successfully reproduced by the in silico MD simulation. By analyzing the structure of the sRII-HtrII complex, the following findings about the photocycle of sRII were obtained: (1) The hydrogen bonds between helixF and other helices determine the direction of the movement of helixF; (2) three amino acids (Arg162, Thr189, Tyr199) are essential for sRII-HtrII binding and contribute to the motion transfer from sRII to HtrII; (3) after the isomerization of retinal, a major conformational change of retinal was caused by proton transfer from Schiff base to Asp75, which, in turn, triggers the steric collision of retinal with Trp171. This is the main reason for the movement of the cytoplasmic side of helixF.
Figures









References
-
- Becke, A.D. 1993. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98 5648–5652.
-
- Case, D.A., Pearlman, D.A., Caldwell, J.W., Cheatham III, T.E., Wang, J., Ross, W.S., Simmerling, C.L., Darden, T.A., Merz, K.M., Standon, R.V., et al. 2002. AMBER7. University of California, San Francisco, CA.
-
- Cieplak, P., Cornell, W.D., Bayly, C., and Kollman, P.A. 1995. Application to the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA and proteins. J. Computat. Chem. 16 1357–1377.
-
- Edman, K., Royant, A., Nollert, P., Maxwell, C.A., Pebay-Peyroula, E., Navarro, J., Neutze, R., and Landau, E.M. 2002. Early structural rearrengements in the photocycle of an integral membrane sensory receptor. Structure 10 473–482. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
