

Multiple synaptic and membrane sites of anesthetic action in the CA1 region of rat hippocampal slices
- PMID: 15579203
- PMCID: PMC543467
- DOI: 10.1186/1471-2202-5-52
Multiple synaptic and membrane sites of anesthetic action in the CA1 region of rat hippocampal slices
Abstract
Background: Anesthesia is produced by a depression of central nervous system function, however, the sites and mechanisms of action underlying this depression remain poorly defined. The present study compared and contrasted effects produced by five general anesthetics on synaptic circuitry in the CA1 region of hippocampal slices.
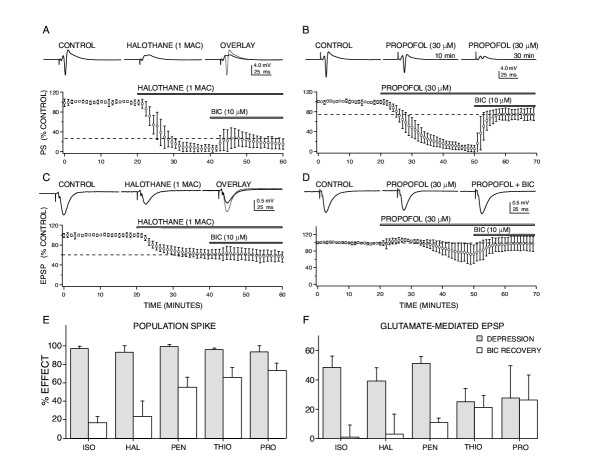
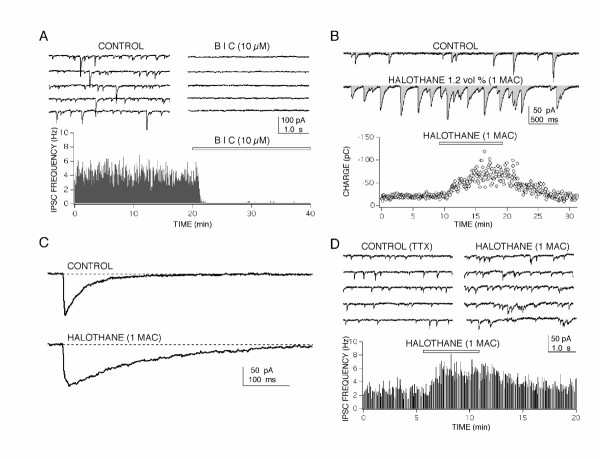
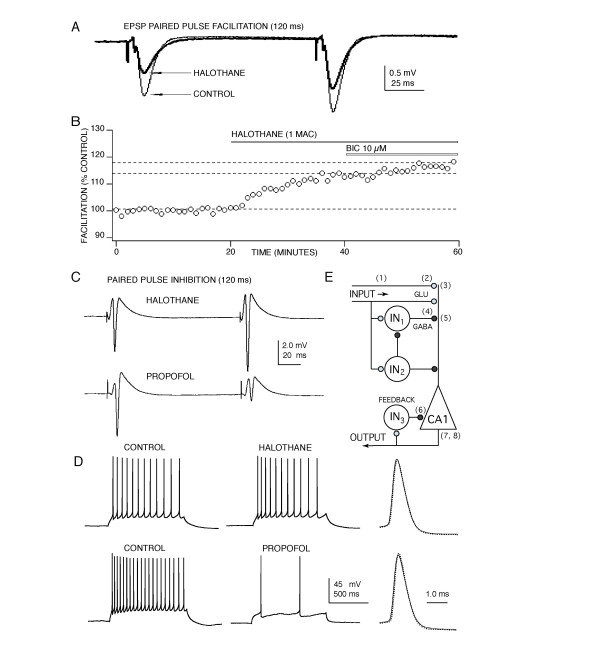
Results: At clinically relevant and equi-effective concentrations, presynaptic and postsynaptic anesthetic actions were evident at glutamate-mediated excitatory synapses and at GABA-mediated inhibitory synapses. In addition, depressant effects on membrane excitability were observed for CA1 neuron discharge in response to direct current depolarization. Combined actions at several of these sites contributed to CA1 circuit depression, but the relative degree of effect at each site was different for each anesthetic studied. For example, most of propofol's depressant effect (> 70 %) was reversed with a GABA antagonist, but only a minor portion of isoflurane's depression was reversed (< 20 %). Differences were also apparent on glutamate synapses-pentobarbital depressed transmission by > 50 %, but thiopental by only < 25 %.
Conclusions: These results, in as much as they may be relevant to anesthesia, indicate that general anesthetics act at several discrete sites, supporting a multi-site, agent specific theory for anesthetic actions. No single effect site (e.g. GABA synapses) or mechanism of action (e.g. depressed membrane excitability) could account for all of the effects produced for any anesthetic studied.
Figures
References
-
- Nicoll RA, Eccles JC, Oshima T, Rubia F. Prolongation of hippocampal inhibitory postsynaptic potentials by barbiturates. Nature. 1975;258:625–627. - PubMed
-
- Tanelian DL, Kosek P, Mody I, Maclver MB. The role of the GABAA receptor/chloride channel complex in anesthesia. Anesthesiology. 1993;78:757–776. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
