

Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice
- PMID: 15579309
- PMCID: PMC2894977
- DOI: 10.1016/j.matbio.2004.09.007
Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice
Abstract
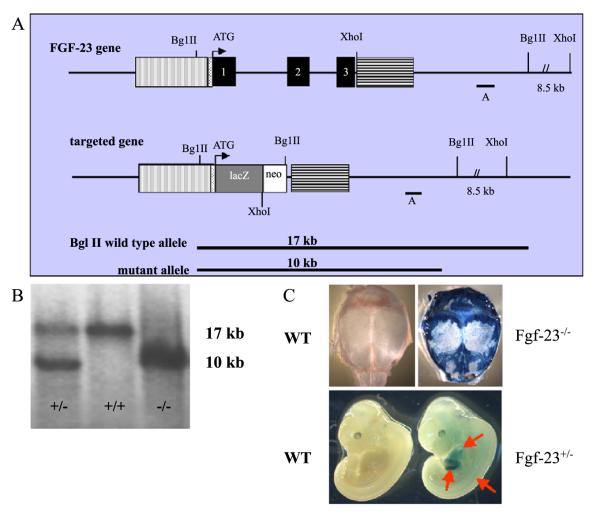
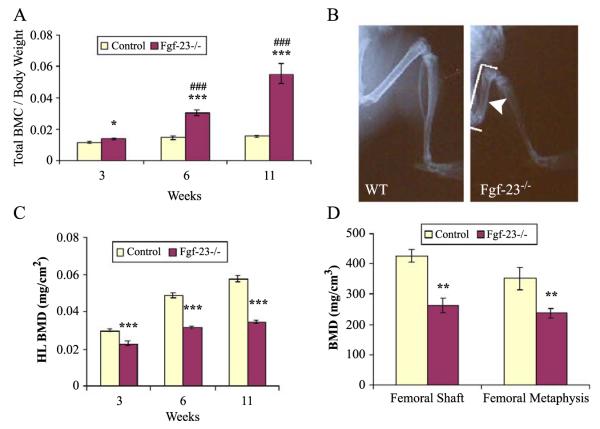
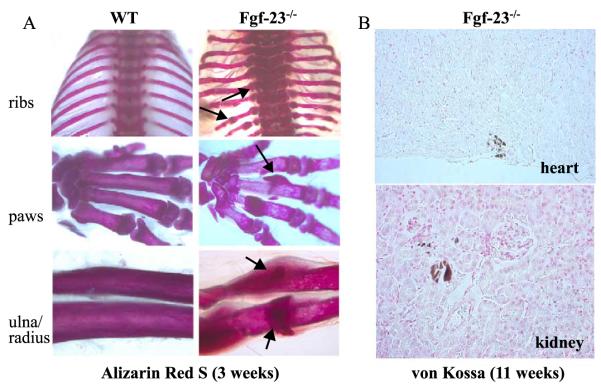
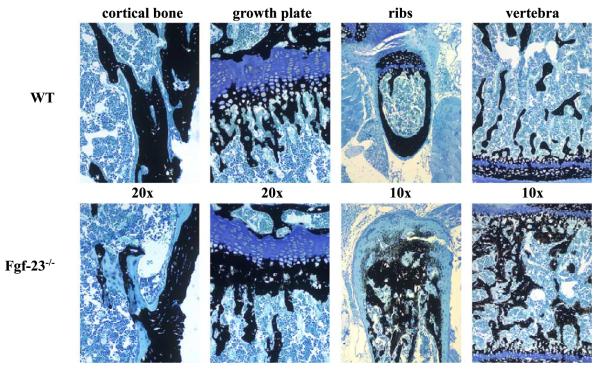
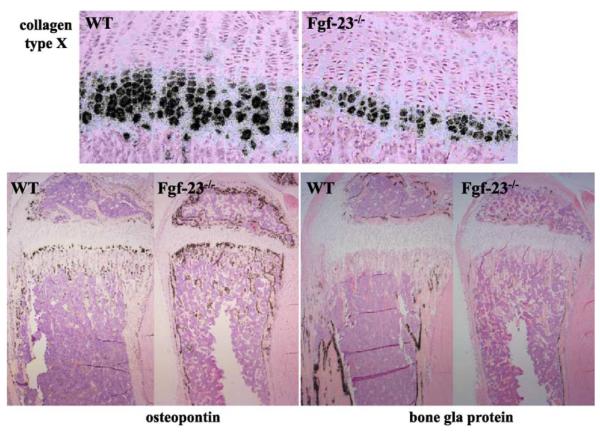
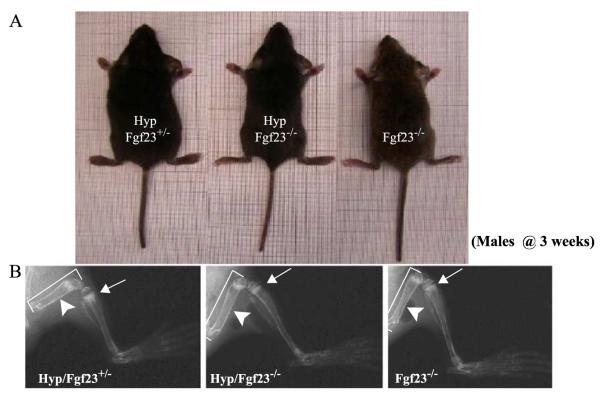
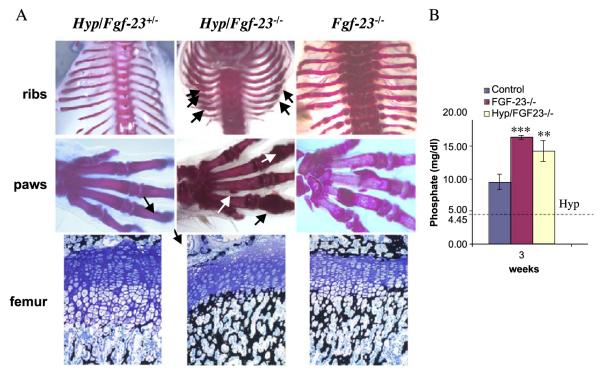
Fibroblast growth factor-23 (FGF-23), a recently identified molecule that is mutated in patients with autosomal dominant hypophosphatemic rickets (ADHR), appears to be involved in the regulation of phosphate homeostasis. Although increased levels of circulating FGF-23 were detected in patients with different phosphate-wasting disorders such as oncogenic osteomalacia (OOM) and X-linked hypophosphatemia (XLH), it is not yet clear whether FGF-23 is directly responsible for the abnormal regulation of mineral ion homeostasis and consequently bone development. To address some of these unresolved questions, we generated a mouse model, in which the entire Fgf-23 gene was replaced with the lacZ gene. Fgf-23 null (Fgf-23-/-) mice showed signs of growth retardation by day 17, developed severe hyperphosphatemia with elevated serum 1,25(OH)2D3 levels, and died by 13 weeks of age. Hyperphosphatemia in Fgf-23-/- mice was accompanied by skeletal abnormalities, as demonstrated by histological, molecular, and various other morphometric analyses. Fgf-23-/-) mice had increased total-body bone mineral content (BMC) but decreased bone mineral density (BMD) of the limbs. Overall, Fgf-23-/- mice exhibited increased mineralization, but also accumulation of unmineralized osteoid leading to marked limb deformities. Moreover, Fgf-23-/- mice showed excessive mineralization in soft tissues, including heart and kidney. To further expand our understanding regarding the role of Fgf-23 in phosphate homeostasis and skeletal mineralization, we crossed Fgf-23-/- animals with Hyp mice, the murine equivalent of XLH. Interestingly, Hyp males lacking both Fgf-23 alleles were indistinguishable from Fgf-23/-/ mice, both in terms of serum phosphate levels and skeletal changes, suggesting that Fgf-23 is upstream of the phosphate regulating gene with homologies to endopeptidases on the X chromosome (Phex) and that the increased plasma Fgf-23 levels in Hyp mice (and in XLH patients) may be at least partially responsible for the phosphate imbalance in this disorder.
Figures
References
-
- ADHR_Consortium Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. The ADHR Consortium. Nat. Genet. 2000;26:345–348. - PubMed
-
- Alfrey AC, LeGendre GR, Kaehny WD. The dialysis encephalopathy syndrome. Possible aluminum intoxication. N. Engl. J. Med. 1976;294:184–188. - PubMed
-
- Aono Y, Shimada T, Yamaziki Y, Hino R, Takeuchi Y, Fujita T, Fukumoto S, Nagano N, Wada M, Yamashita T. The neutralization of FGF-23 ameliorates hypophosphatemia and rickets in Hyp mice. J. Bone Miner. Res. 2003;18(Suppl. 2):1056.
-
- Argiro L, Desbarats M, Glorieux FH, Ecarot B. Mepe, the gene encoding a tumor-secreted protein in oncogenic hypophosphatemic osteomalacia, is expressed in bone. Genomics. 2001;74:342–351. - PubMed
-
- Bai X, Miao D, Panda D, Grady S, McKee MD, Goltzman D, Karaplis AC. Partial rescue of the Hyp phenotype by osteoblast-targeted PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) expression. Mol. Endocrinol. 2002;16:2913–2925. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
