

Global protein function annotation through mining genome-scale data in yeast Saccharomyces cerevisiae
- PMID: 15585665
- PMCID: PMC535686
- DOI: 10.1093/nar/gkh978
Global protein function annotation through mining genome-scale data in yeast Saccharomyces cerevisiae
Abstract
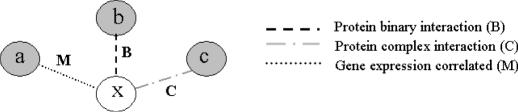
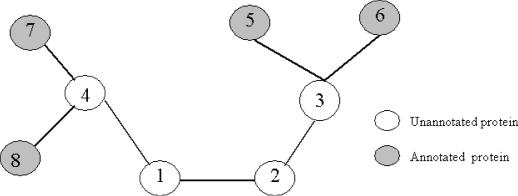
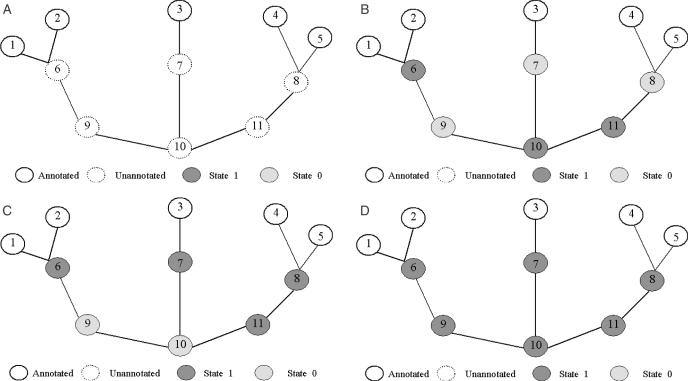
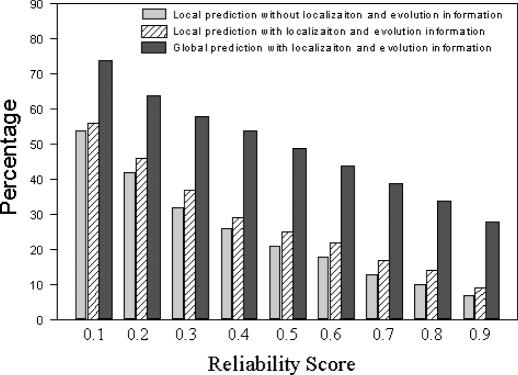
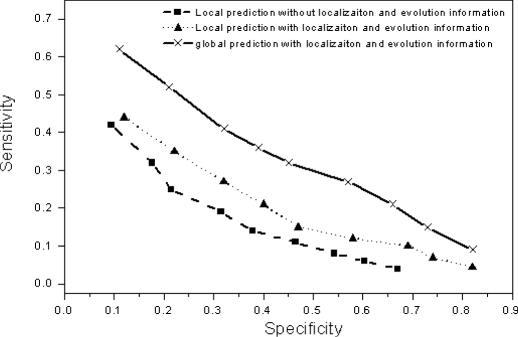
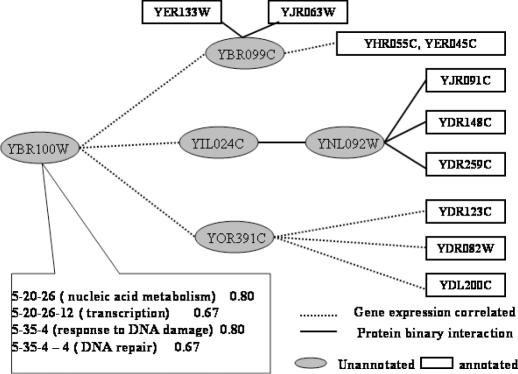
As we are moving into the post genome-sequencing era, various high-throughput experimental techniques have been developed to characterize biological systems on the genomic scale. Discovering new biological knowledge from the high-throughput biological data is a major challenge to bioinformatics today. To address this challenge, we developed a Bayesian statistical method together with Boltzmann machine and simulated annealing for protein functional annotation in the yeast Saccharomyces cerevisiae through integrating various high-throughput biological data, including yeast two-hybrid data, protein complexes and microarray gene expression profiles. In our approach, we quantified the relationship between functional similarity and high-throughput data, and coded the relationship into 'functional linkage graph', where each node represents one protein and the weight of each edge is characterized by the Bayesian probability of function similarity between two proteins. We also integrated the evolution information and protein subcellular localization information into the prediction. Based on our method, 1802 out of 2280 unannotated proteins in yeast were assigned functions systematically.
Figures
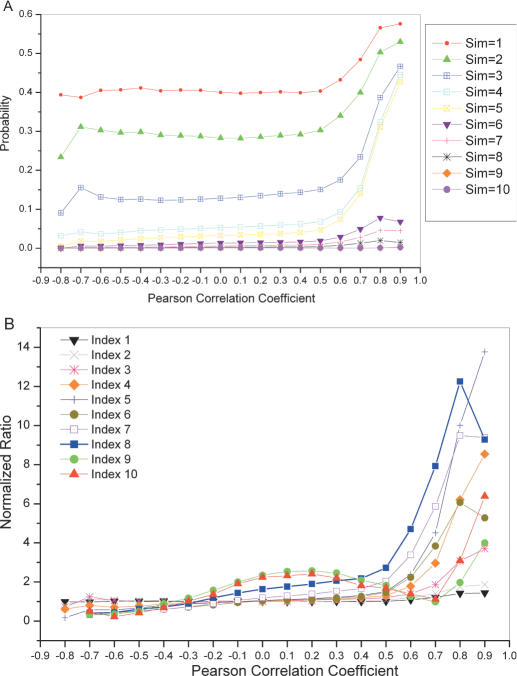
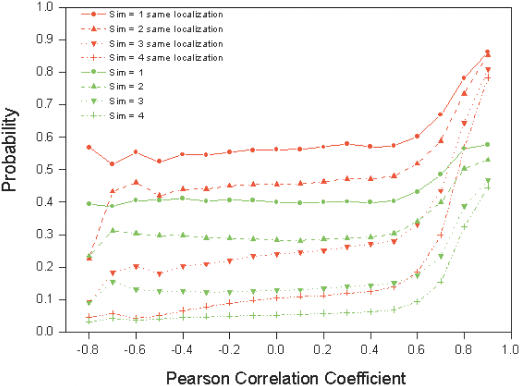
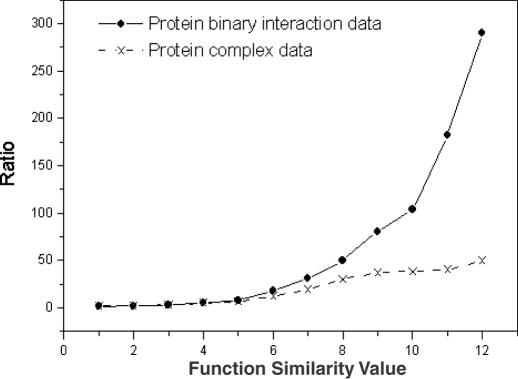
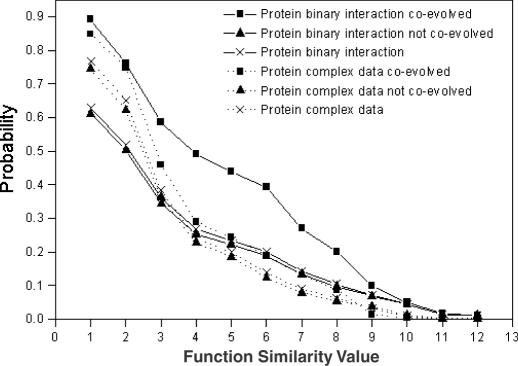

References
-
- Goffeau A., Barrell,B.G., Bussey,H., Davis,R.W., Dujon,B., Feldmann,H., Galibert,F., Hoheisel,J.D., Jacq,C., Johnston,M., Louis,E.J, Mewes,H.W., Murakami,Y., Philippsen,P., Tettelin,H. and Oliver,S.G. (1996) Life with 6000 genes. Science, 546, 346–352. - PubMed
-
- Marcotte E., Pellegrini,M., Thompson,M., Yeates,T. and Eisenberg,D. (1999) A combined algorithm for genome-wide prediction of protein function. Nature, 402, 83–86. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
