

Comparing functional annotation analyses with Catmap
- PMID: 15588298
- PMCID: PMC543458
- DOI: 10.1186/1471-2105-5-193
Comparing functional annotation analyses with Catmap
Abstract
Background: Ranked gene lists from microarray experiments are usually analysed by assigning significance to predefined gene categories, e.g., based on functional annotations. Tools performing such analyses are often restricted to a category score based on a cutoff in the ranked list and a significance calculation based on random gene permutations as null hypothesis.
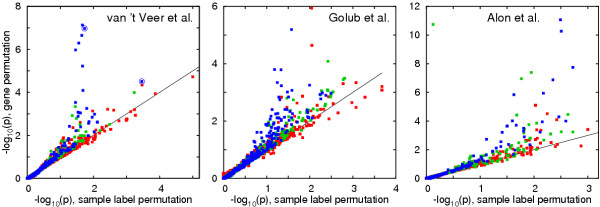
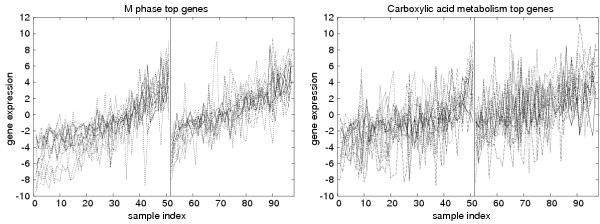
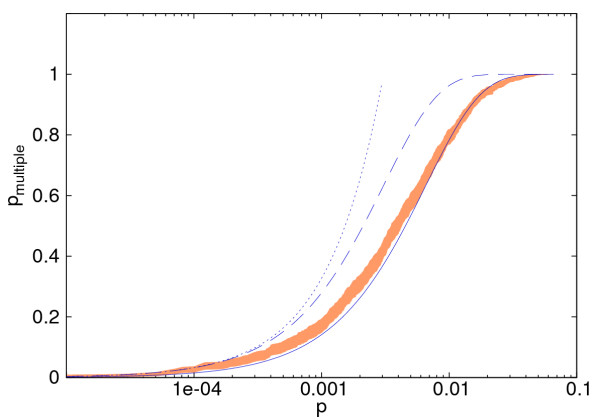
Results: We analysed three publicly available data sets, in each of which samples were divided in two classes and genes ranked according to their correlation to class labels. We developed a program, Catmap (available for download at http://bioinfo.thep.lu.se/Catmap), to compare different scores and null hypotheses in gene category analysis, using Gene Ontology annotations for category definition. When a cutoff-based score was used, results depended strongly on the choice of cutoff, introducing an arbitrariness in the analysis. Comparing results using random gene permutations and random sample permutations, respectively, we found that the assigned significance of a category depended strongly on the choice of null hypothesis. Compared to sample label permutations, gene permutations gave much smaller p-values for large categories with many coexpressed genes.
Conclusions: In gene category analyses of ranked gene lists, a cutoff independent score is preferable. The choice of null hypothesis is very important; random gene permutations does not work well as an approximation to sample label permutations.
Figures
References
-
- Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN. Gominer: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
