

Whole genome DNA copy number changes identified by high density oligonucleotide arrays
- PMID: 15588488
- PMCID: PMC3525261
- DOI: 10.1186/1479-7364-1-4-287
Whole genome DNA copy number changes identified by high density oligonucleotide arrays
Abstract
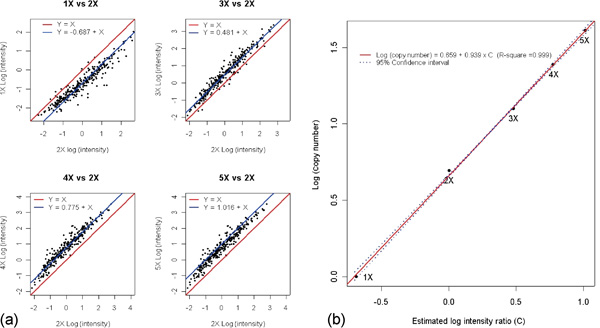
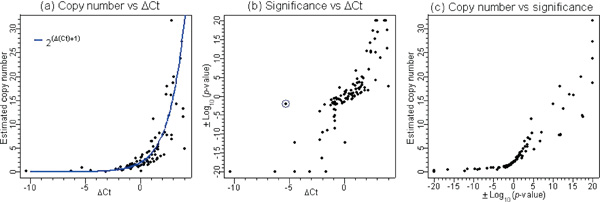
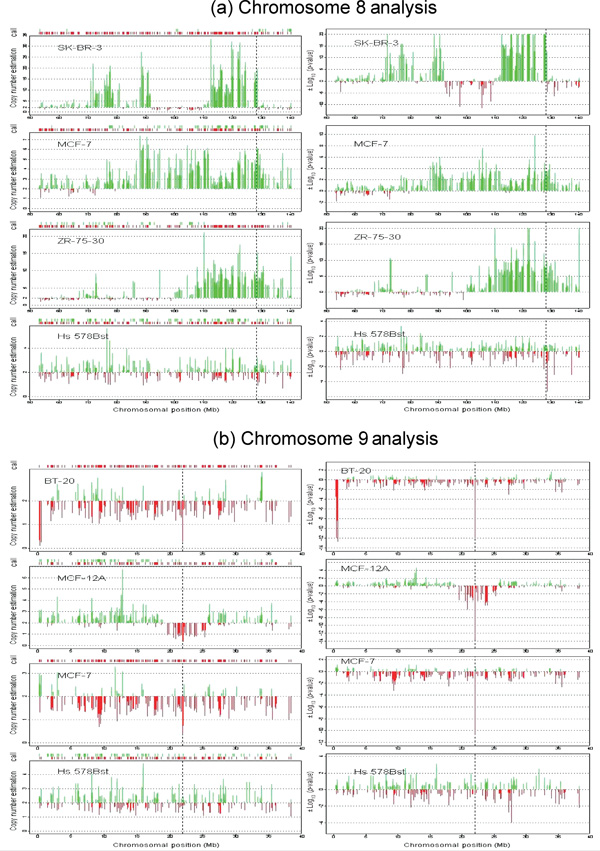
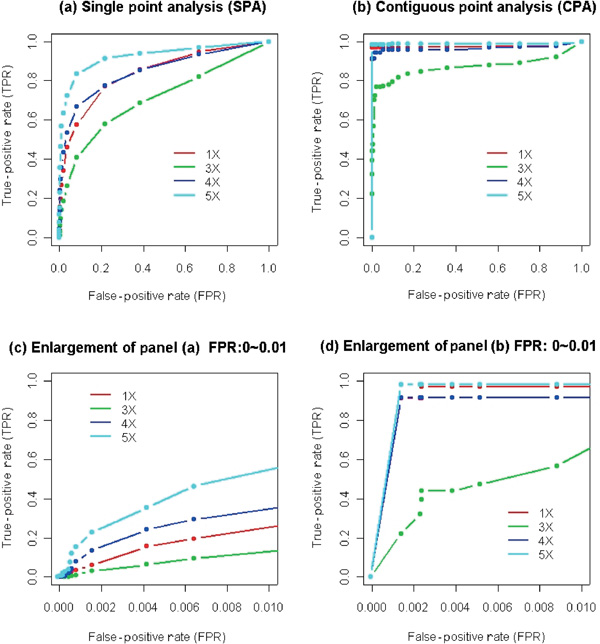
Changes in DNA copy number are one of the hallmarks of the genetic instability common to most human cancers. Previous microarray-based methods have been used to identify chromosomal gains and losses; however, they are unable to genotype alleles at the level of single nucleotide polymorphisms (SNPs). Here we describe a novel algorithm that uses a recently developed high-density oligonucleotide array-based SNP genotyping method, whole genome sampling analysis (WGSA), to identify genome-wide chromosomal gains and losses at high resolution. WGSA simultaneously genotypes over 10,000 SNPs by allele-specific hybridisation to perfect match (PM) and mismatch (MM) probes synthesised on a single array. The copy number algorithm jointly uses PM intensity and discrimination ratios between paired PM and MM intensity values to identify and estimate genetic copy number changes. Values from an experimental sample are compared with SNP-specific distributions derived from a reference set containing over 100 normal individuals to gain statistical power. Genomic regions with statistically significant copy number changes can be identified using both single point analysis and contiguous point analysis of SNP intensities. We identified multiple regions of amplification and deletion using a panel of human breast cancer cell lines. We verified these results using an independent method based on quantitative polymerase chain reaction and found that our approach is both sensitive and specific and can tolerate samples which contain a mixture of both tumour and normal DNA. In addition, by using known allele frequencies from the reference set, statistically significant genomic intervals can be identified containing contiguous stretches of homozygous markers, potentially allowing the detection of regions undergoing loss of heterozygosity (LOH) without the need for a matched normal control sample. The coupling of LOH analysis, via SNP genotyping, with copy number estimations using a single array provides additional insight into the structure of genomic alterations. With mean and median inter-SNP euchromatin distances of 244 kilobases (kb) and 119 kb, respectively, this method affords a resolution that is not easily achievable with non-oligonucleotide-based experimental approaches.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
