

Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area
- PMID: 15590923
- PMCID: PMC4857882
- DOI: 10.1523/JNEUROSCI.3695-04.2004
Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area
Abstract
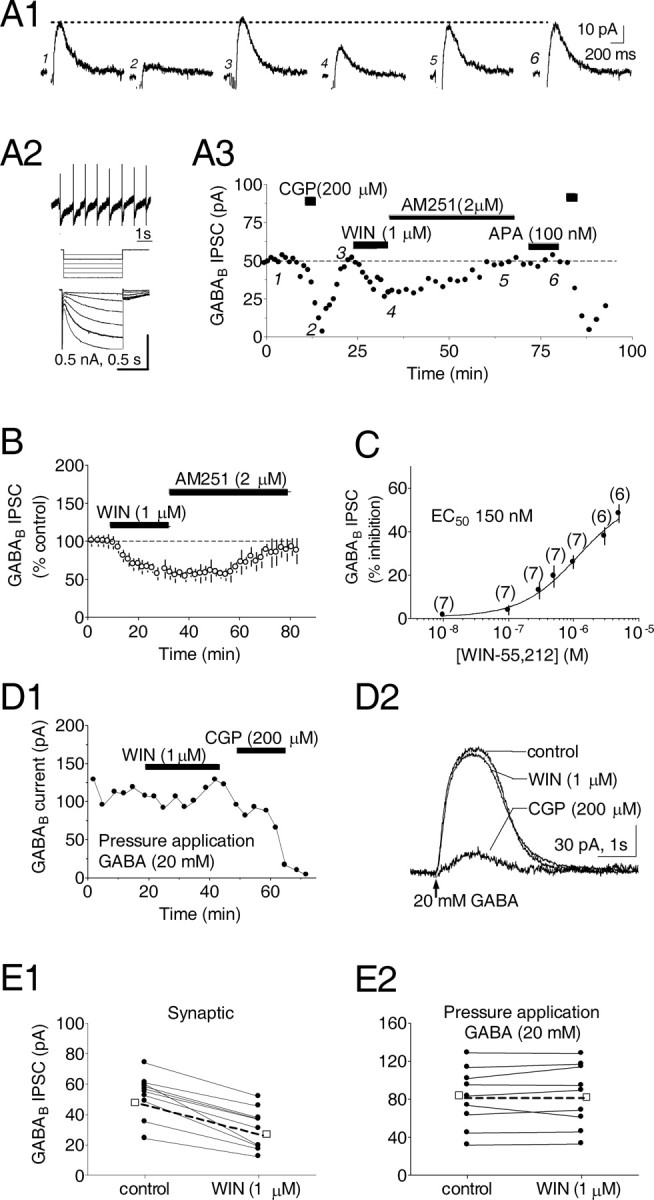
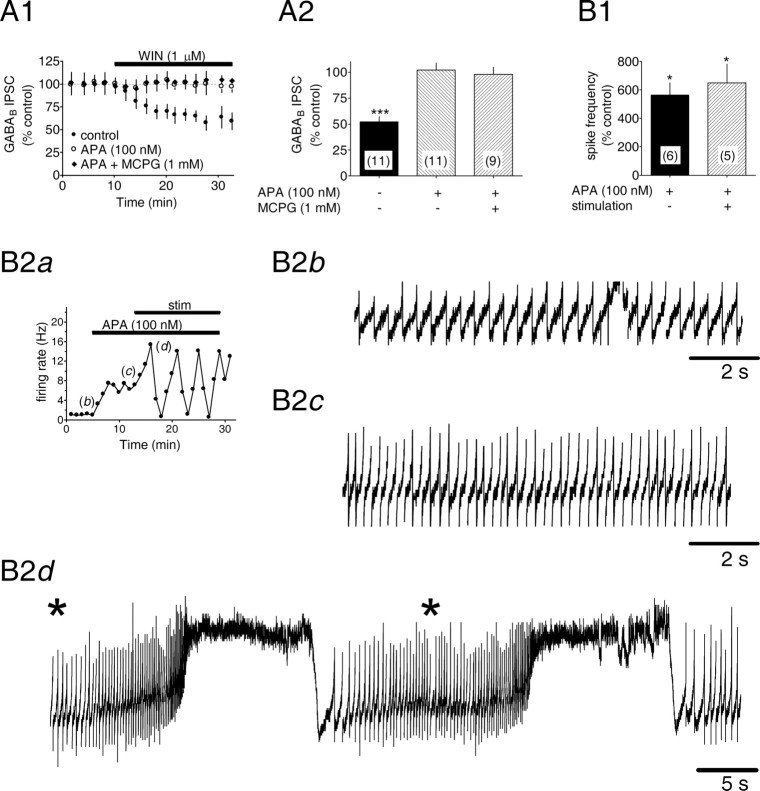
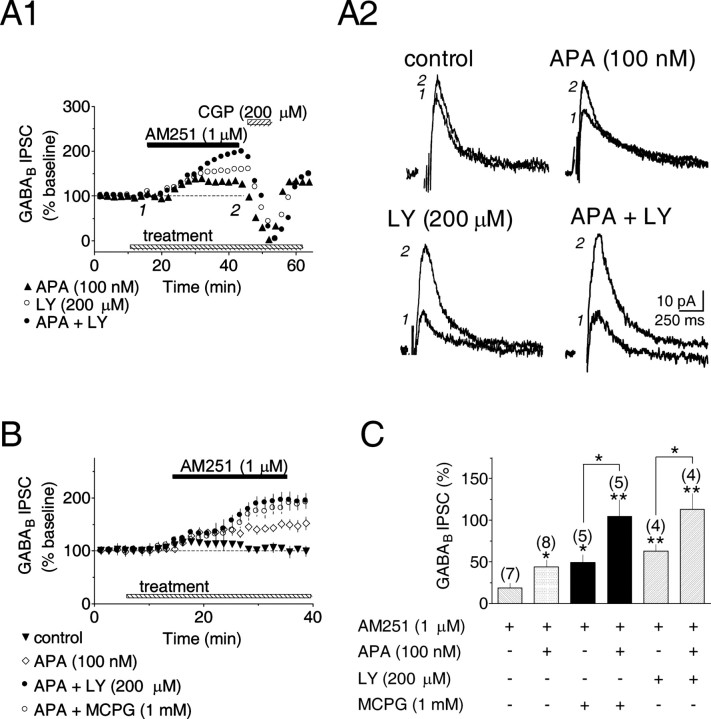
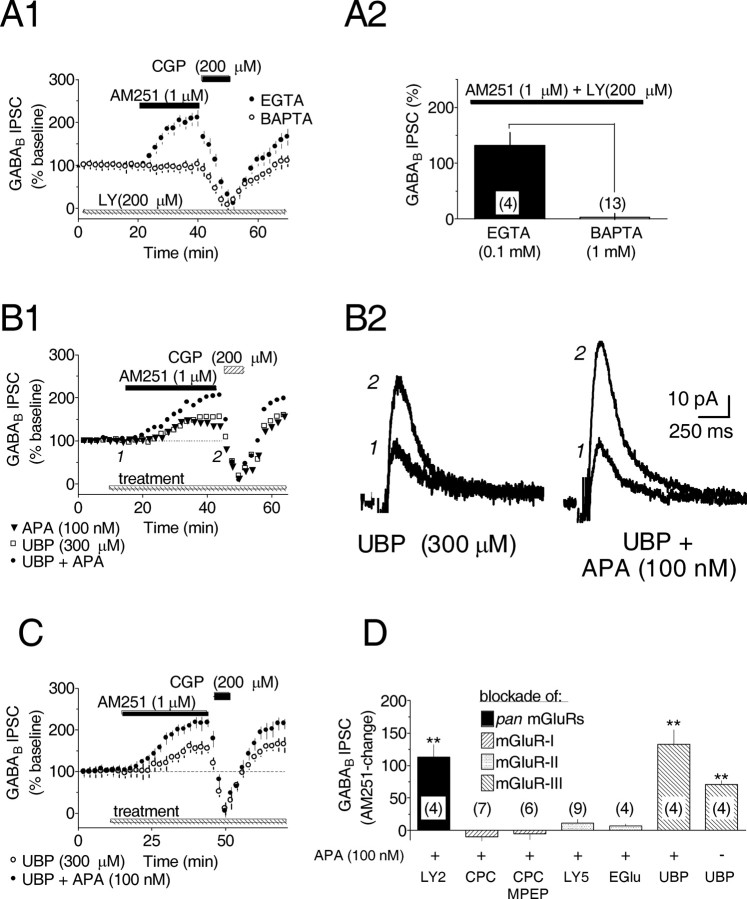
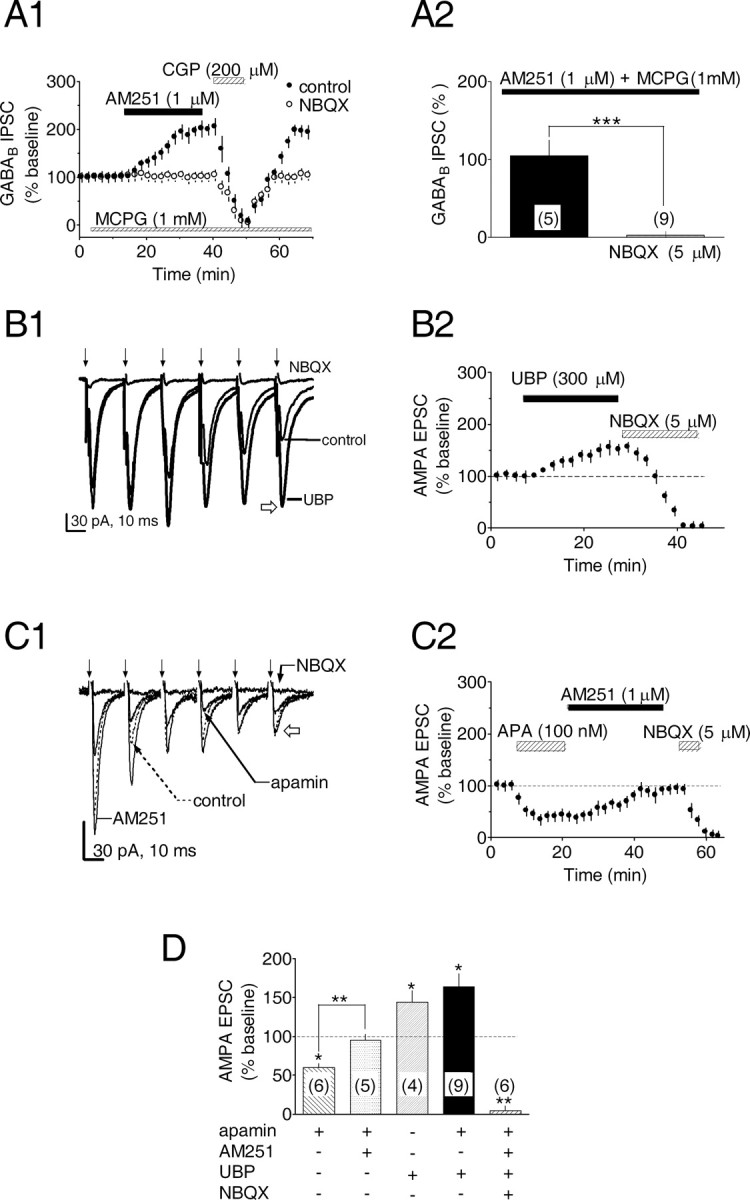
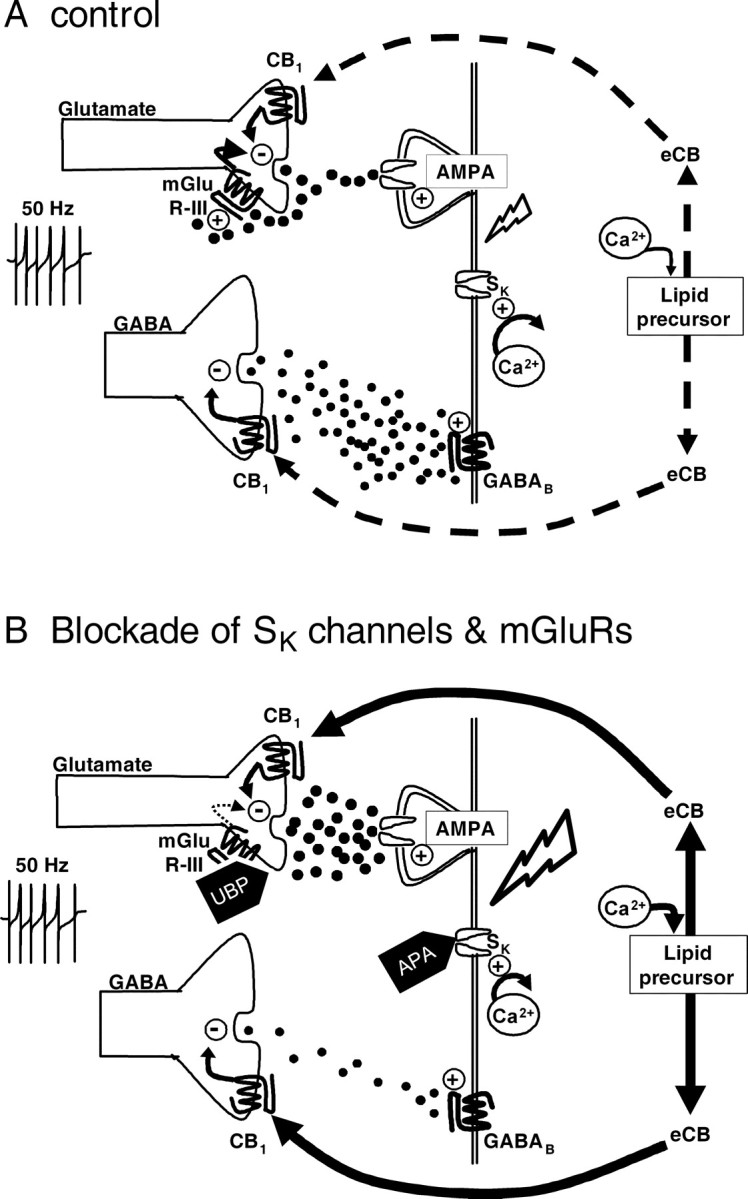
Dopamine (DA) neurons in the ventral tegmental area have been implicated in psychiatric disorders and drug abuse. Understanding the mechanisms through which their activity is regulated via the modulation of afferent input is imperative to understanding their roles in these conditions. Here we demonstrate that endocannabinoids liberated from DA neurons activate cannabinoid CB1 receptors located on glutamatergic axons and on GABAergic terminals targeting GABA(B) receptors located on these cells. Endocannabinoid release was initiated by inhibiting either presynaptic type-III metabotropic glutamate receptors or postsynaptic calcium-activated potassium channels, two conditions that also promote enhanced DA neuron excitability and bursting. Thus, activity-dependent release of endocannabinoids may act as a regulatory feedback mechanism to inhibit synaptic inputs in response to DA neuron bursting, thereby regulating firing patterns that may fine-tune DA release from afferent terminals.
Figures
References
-
- Alger BE (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286. - PubMed
-
- Bonci A, Grillner P, Siniscalchi A, Mercuri NB, Bernardi G (1997) Glutamate metabotropic receptor agonists depress excitatory and inhibitory transmission on rat mesencephalic principal neurons. Eur J Neurosci 9: 2359-2369. - PubMed
-
- Cameron DL, Wessendorf MW, Williams JT (1997) A subset of ventral tegmental area neurons is inhibited by dopamine, 5-hydroxytryptamine and opioids. Neuroscience 77: 155-166. - PubMed
-
- Charara A, Smith Y, Parent A (1996) Glutamatergic inputs from the pedunculopontine nucleus to midbrain dopaminergic neurons in primates: Phaseolus vulgaris-leucoagglutinin anterograde labeling combined with postembedding glutamate and GABA immunohistochemistry. J Comp Neurol 364: 254-266. - PubMed
-
- Cheer JF, Kendall DA, Mason R, Marsden CA (2003) Differential cannabinoid-induced electrophysiological effects in rat ventral tegmentum. Neuropharmacology 44: 633-641. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources