

Advances in protein complex analysis using mass spectrometry
- PMID: 15611014
- PMCID: PMC1665575
- DOI: 10.1113/jphysiol.2004.080440
Advances in protein complex analysis using mass spectrometry
Abstract
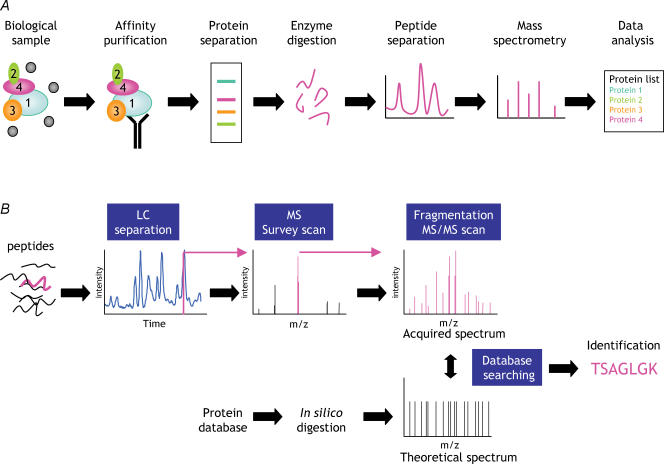
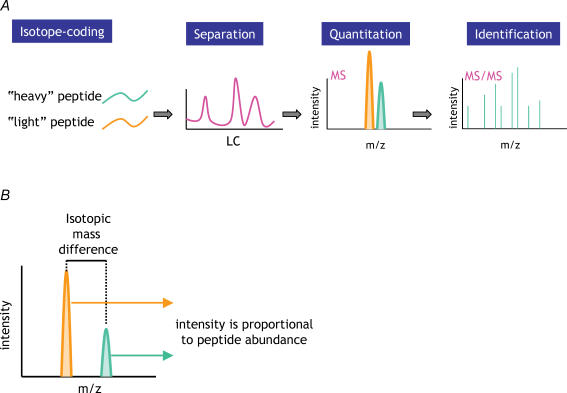
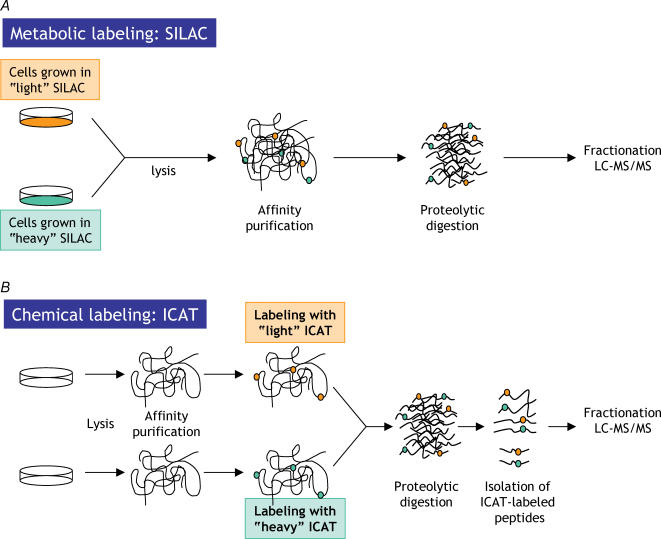
Proteins often function as components of larger complexes to perform a specific function, and formation of these complexes may be regulated. For example, intracellular signalling events often require transient and/or regulated protein-protein interactions for propagation, and protein binding to a specific DNA sequence, RNA molecule or metabolite is often regulated to modulate a particular cellular function. Thus, characterizing protein complexes can offer important insights into protein function. This review describes recent important advances in mass spectrometry (MS)-based techniques for the analysis of protein complexes. Following brief descriptions of how proteins are identified using MS, and general protein complex purification approaches, we address two of the most important issues in these types of studies: specificity and background protein contaminants. Two basic strategies for increasing specificity and decreasing background are presented: whereas (1) tandem affinity purification (TAP) of tagged proteins of interest can dramatically improve the signal-to-noise ratio via the generation of cleaner samples, (2) stable isotopic labelling of proteins may be used to discriminate between contaminants and bona fide binding partners using quantitative MS techniques. Examples, as well as advantages and disadvantages of each approach, are presented.
Figures


References
-
- Bauer A, Kuster B. Affinity purification-mass spectrometry. Powerful tools for the characterization of protein complexes. Eur J Biochem. 2003;270:570–578. - PubMed
-
- Blagoev B, Kratchmarova I, Ong SE, Nielsen M, Foster LJ, Mann M. A proteomics strategy to elucidate functional protein–protein interactions applied to EGF signaling. Nat Biotechnol. 2003;21:315–318. - PubMed
-
- Blagoev B, Ong SE, Kratchmarova I, Mann M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat Biotechnol. 2004;22:1139–1145. - PubMed
-
- Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous