

Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome
- PMID: 15611184
- PMCID: PMC1448716
- DOI: 10.1534/genetics.104.032375
Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome
Abstract
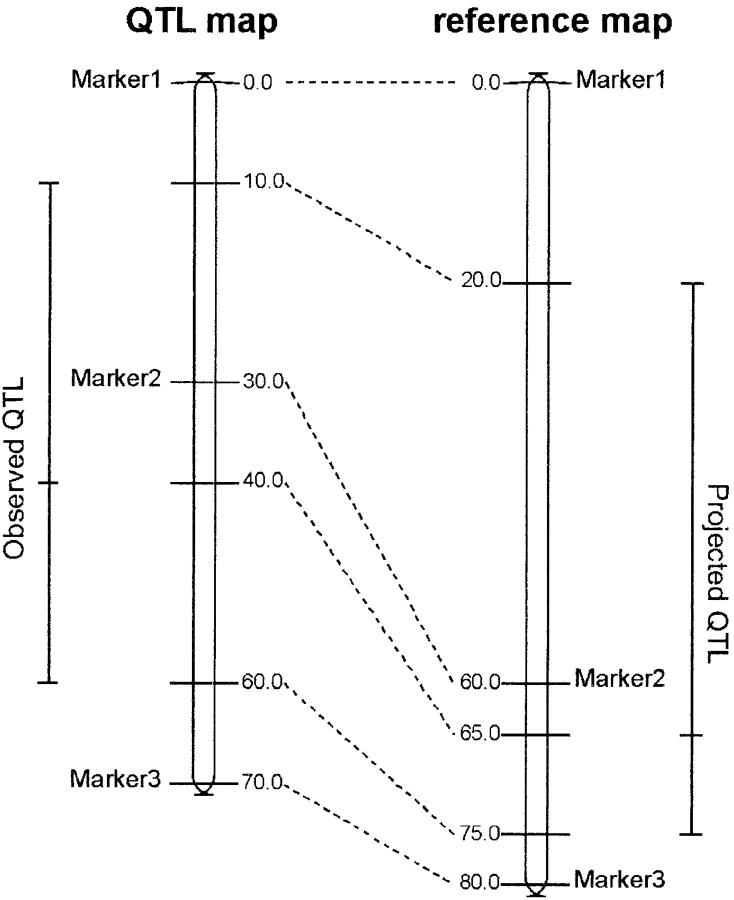
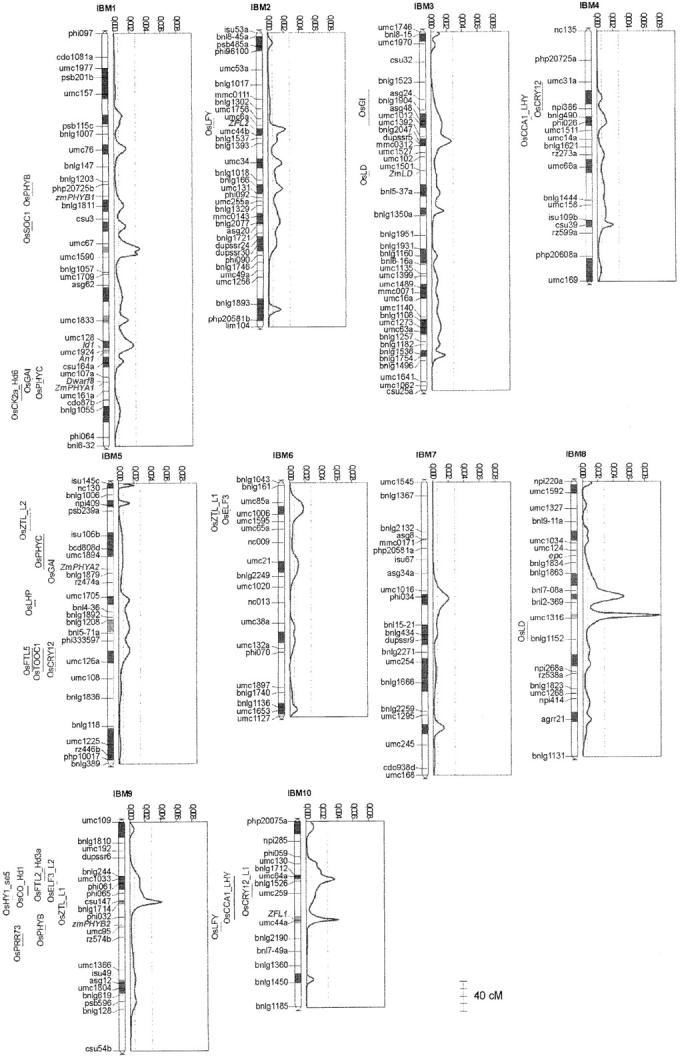
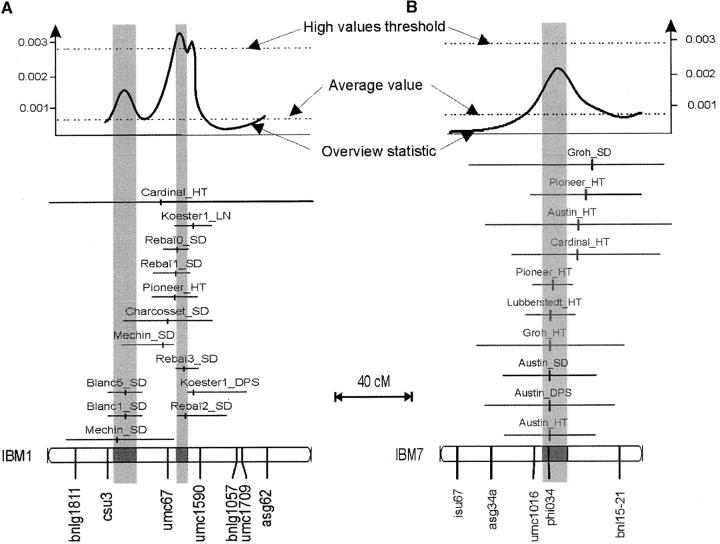
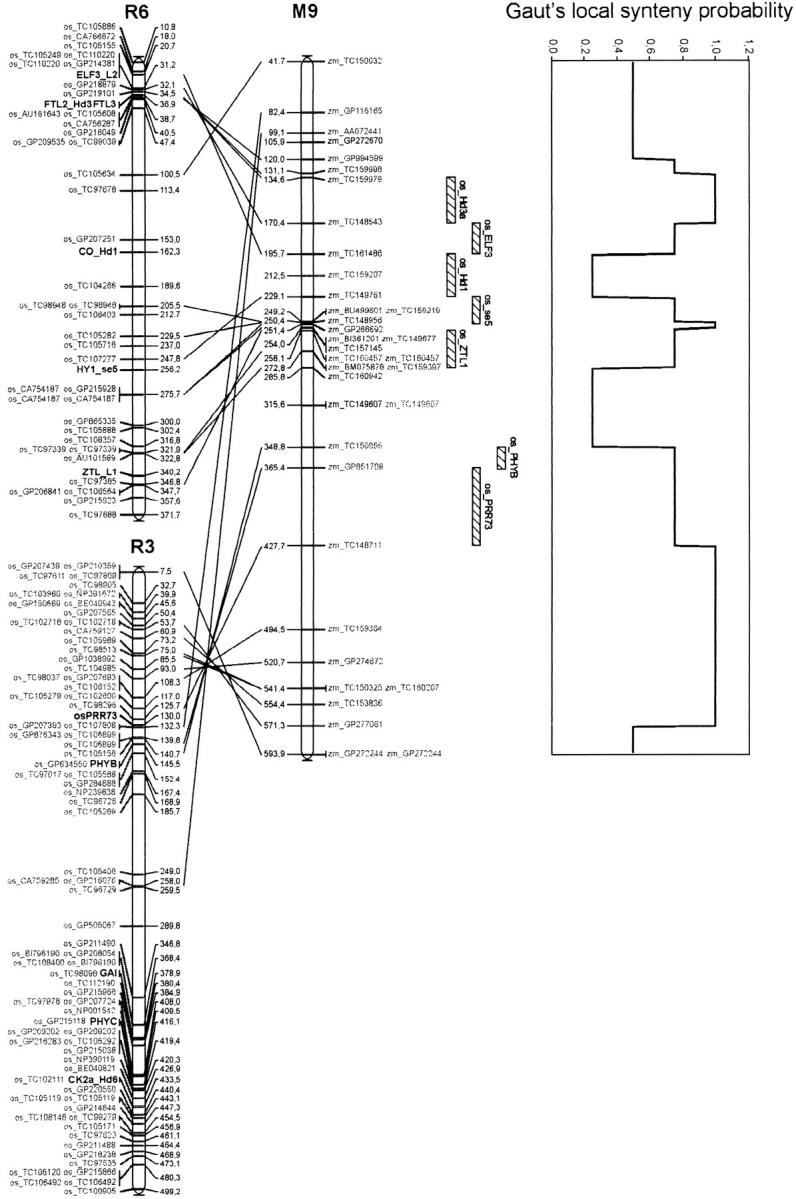
Genetic architecture of flowering time in maize was addressed by synthesizing a total of 313 quantitative trait loci (QTL) available for this trait. These were analyzed first with an overview statistic that highlighted regions of key importance and then with a meta-analysis method that yielded a synthetic genetic model with 62 consensus QTL. Six of these displayed a major effect. Meta-analysis led in this case to a twofold increase in the precision in QTL position estimation, when compared to the most precise initial QTL position within the corresponding region. The 62 consensus QTL were compared first to the positions of the few flowering-time candidate genes that have been mapped in maize. We then projected rice candidate genes onto the maize genome using a synteny conservation approach based on comparative mapping between the maize genetic map and japonica rice physical map. This yielded 19 associations between maize QTL and genes involved in flowering time in rice and in Arabidopsis. Results suggest that the combination of meta-analysis within a species of interest and synteny-based projections from a related model plant can be an efficient strategy for identifying new candidate genes for trait variation.
Figures

References
-
- Abler, B. S. B., M. D. Edwards and C. W. Stuber, 1991. Isoenzymatic identification of quantitative trait loci in crosses of elite maize inbreds. Crop Sci. 31: 267–274.
-
- Arcade, A., A. Labourdette, M. Falque, B. Mangin, F. Chardon et al., 2004. BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20: 2324–2326. - PubMed
-
- Austin, D. F., and M. Lee, 1996. Genetic resolution and verification of quantitative trait loci for flowering and plant height with recombinant inbred lines of maize. Genome 39: 957–968. - PubMed
-
- Basu, D., K. Dehesh, H. J. Schneider-Poetsch, S. E. Harrington, S. R. McCouch et al., 2000. Rice PHYC gene: structure, expression, map position and evolution. Plant Mol. Biol. 41: 27–42. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
