

An empirical analysis of training protocols for probabilistic gene finders
- PMID: 15613242
- PMCID: PMC544851
- DOI: 10.1186/1471-2105-5-206
An empirical analysis of training protocols for probabilistic gene finders
Erratum in
- BMC Bioinformatics. 2005;6:193
Abstract
Background: Generalized hidden Markov models (GHMMs) appear to be approaching acceptance as a de facto standard for state-of-the-art ab initio gene finding, as evidenced by the recent proliferation of GHMM implementations. While prevailing methods for modeling and parsing genes using GHMMs have been described in the literature, little attention has been paid as of yet to their proper training. The few hints available in the literature together with anecdotal observations suggest that most practitioners perform maximum likelihood parameter estimation only at the local submodel level, and then attend to the optimization of global parameter structure using some form of ad hoc manual tuning of individual parameters.
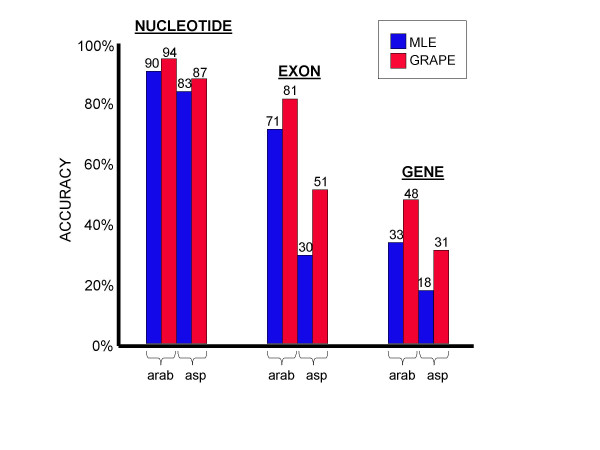
Results: We decided to investigate the utility of applying a more systematic optimization approach to the tuning of global parameter structure by implementing a global discriminative training procedure for our GHMM-based gene finder. Our results show that significant improvement in prediction accuracy can be achieved by this method.
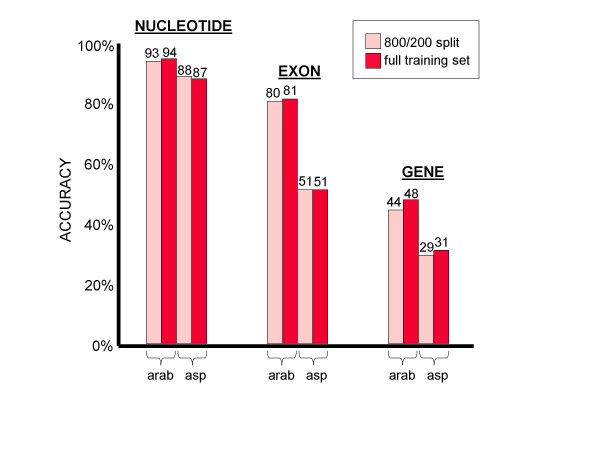
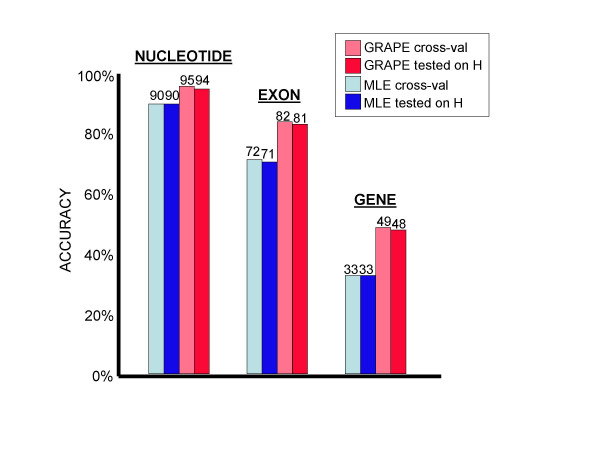
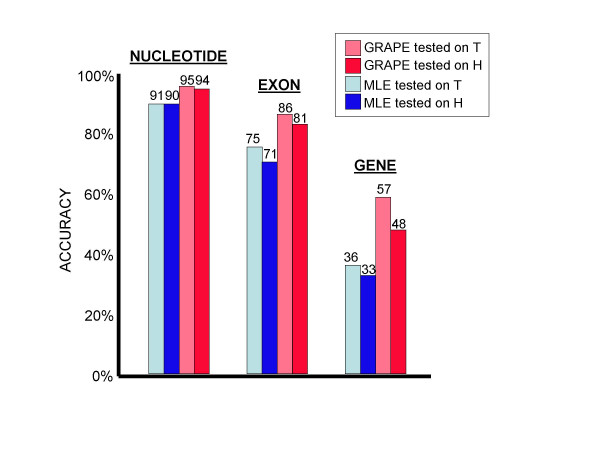
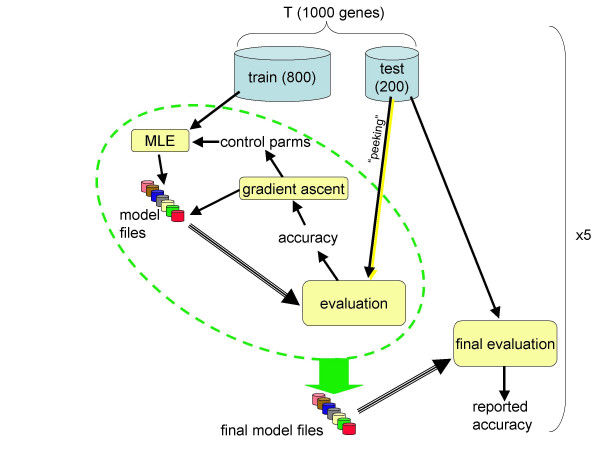
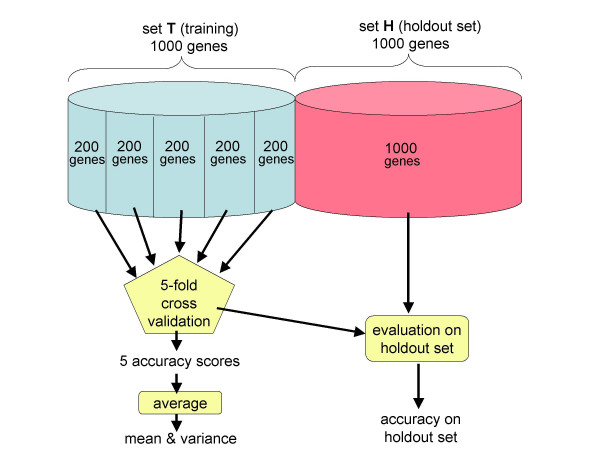
Conclusions: We conclude that training of GHMM-based gene finders is best performed using some form of discriminative training rather than simple maximum likelihood estimation at the submodel level, and that generalized gradient ascent methods are suitable for this task. We also conclude that partitioning of training data for the twin purposes of maximum likelihood initialization and gradient ascent optimization appears to be unnecessary, but that strict segregation of test data must be enforced during final gene finder evaluation to avoid artificially inflated accuracy measurements.
Figures
References
-
- Kulp D, Haussler D, Reese MG, Eeckman FH. A generalized hidden Markov model for the recognition of human genes in DNA. In: States DJ, Agarwal P, Gaasterland T, Hunter L, Smith RF, editor. In Proceedings of the Fourth International Conference on Intelligent Systems for Molecular Biology: 12–15 June 1996 St Louis. Menlo Park: American Association for Artificial Intelligence; 1996. pp. 134–142. - PubMed
-
- Burge C. PhD thesis. Stanford University, Mathematics Department; 1997. Identification of genes in human genomic DNA.
-
- Cawley SE, Wirth AI, Speed TP. Phat – a gene finding program for Plasmodium falciparum. Mol Biochem Parasitol. 2001;118:167–174. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
