

Involvement of influenza virus PA subunit in assembly of functional RNA polymerase complexes
- PMID: 15613301
- PMCID: PMC538542
- DOI: 10.1128/JVI.79.2.732-744.2005
Involvement of influenza virus PA subunit in assembly of functional RNA polymerase complexes
Abstract
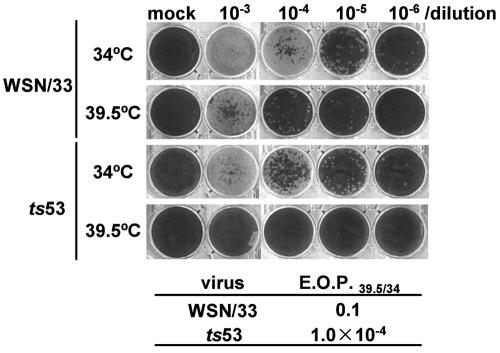
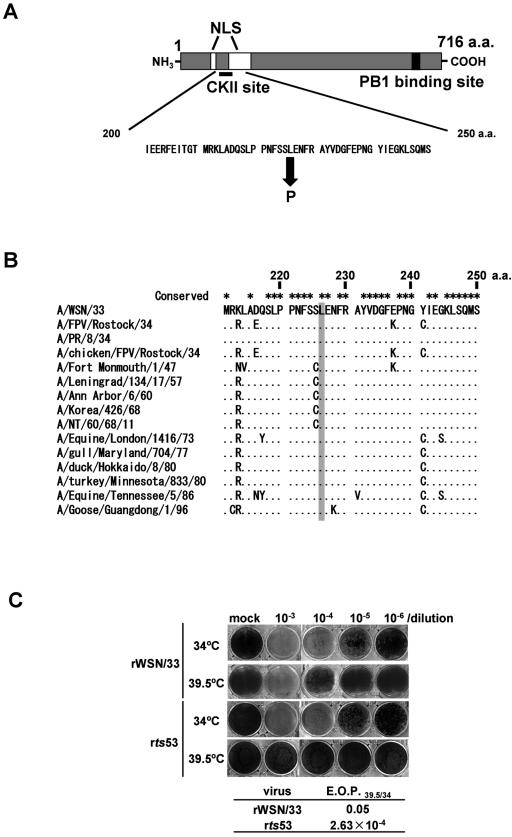
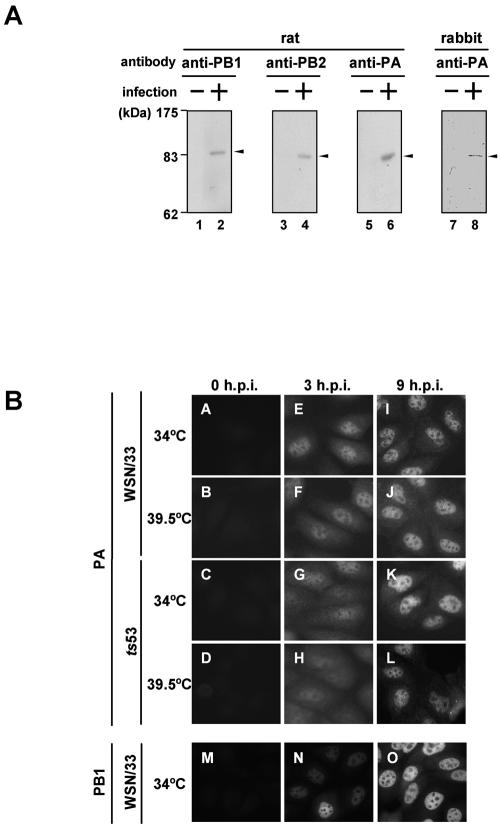
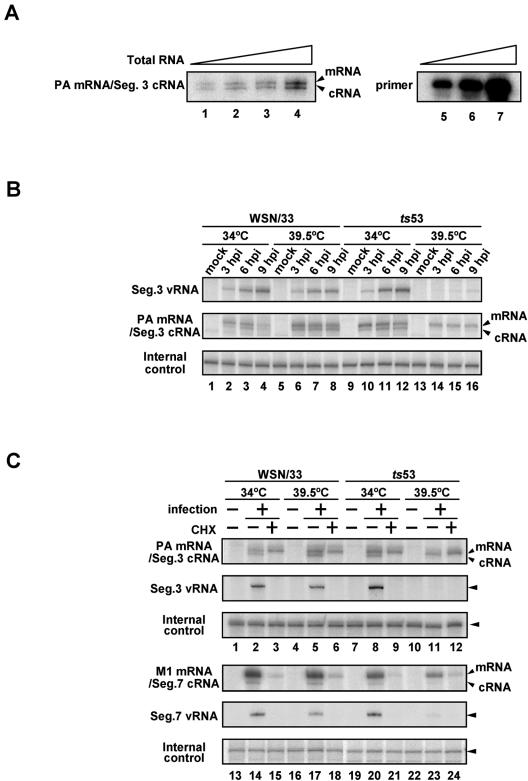
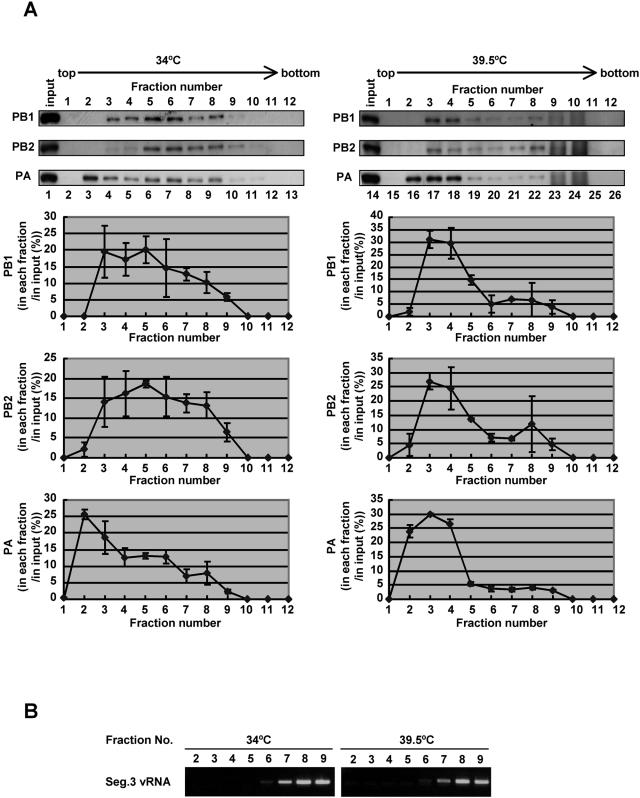
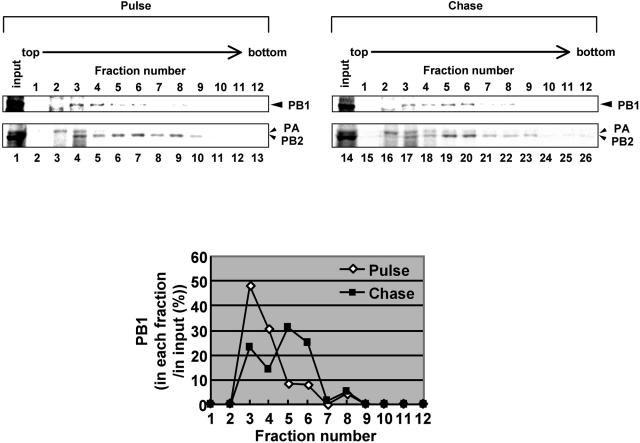
The RNA-dependent RNA polymerase of influenza virus consists of three subunits, PB1, PB2, and PA, and synthesizes three kinds of viral RNAs, vRNA, cRNA, and mRNA. PB1 is a catalytic subunit; PB2 recognizes the cap structure for generation of the primer for transcription; and PA is thought to be involved in viral RNA replication. However, the process of polymerase complex assembly and the exact nature of polymerase complexes involved in synthesis of the three different RNA species are not yet clear. ts53 virus is a temperature-sensitive (ts) mutant derived from A/WSN/33 (A. Sugiura, M. Ueda, K. Tobita, and C. Enomoto, Virology 65:363-373, 1975). We confirmed that the mRNA synthesis level of ts53 remains unaffected at the nonpermissive temperature, whereas vRNA synthesis is largely reduced. Sequencing of the gene encoding ts53 PA and recombinant virus rescue experiments revealed that an amino acid change from Leu to Pro at amino acid position 226 is causative of temperature sensitivity. By glycerol density gradient analyses of nuclear extracts prepared from wild-type virus-infected cells, we found that polymerase proteins sediment in three fractions: one (H fraction) consists of RNP complexes, another (M fraction) contains active polymerases but not viral RNA, and the other (L fraction) contains inactive forms of polymerases. Pulse-chase experiments showed that polymerases in the L fraction are converted to those in the M fraction. In ts53-infected cells, polymerases accumulated in the L fraction. These results strongly suggest that PA is involved in the assembly of functional viral RNA polymerase complexes from their inactive intermediates.
Figures

References
-
- Braam, J., I. Ulmanen, and R. M. Krug. 1983. Molecular model of a eucaryotic transcription complex: functions and movements of influenza P proteins during capped RNA-primed transcription. Cell 34:609-618. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
