

Solution structure of choline binding protein A, the major adhesin of Streptococcus pneumoniae
- PMID: 15616594
- PMCID: PMC544903
- DOI: 10.1038/sj.emboj.7600490
Solution structure of choline binding protein A, the major adhesin of Streptococcus pneumoniae
Abstract
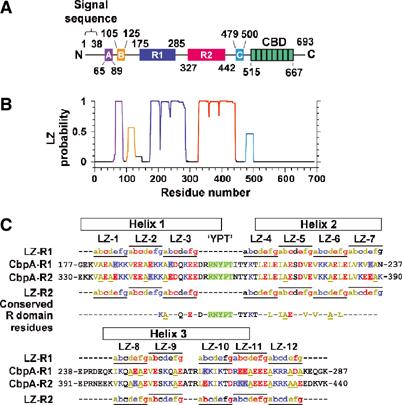
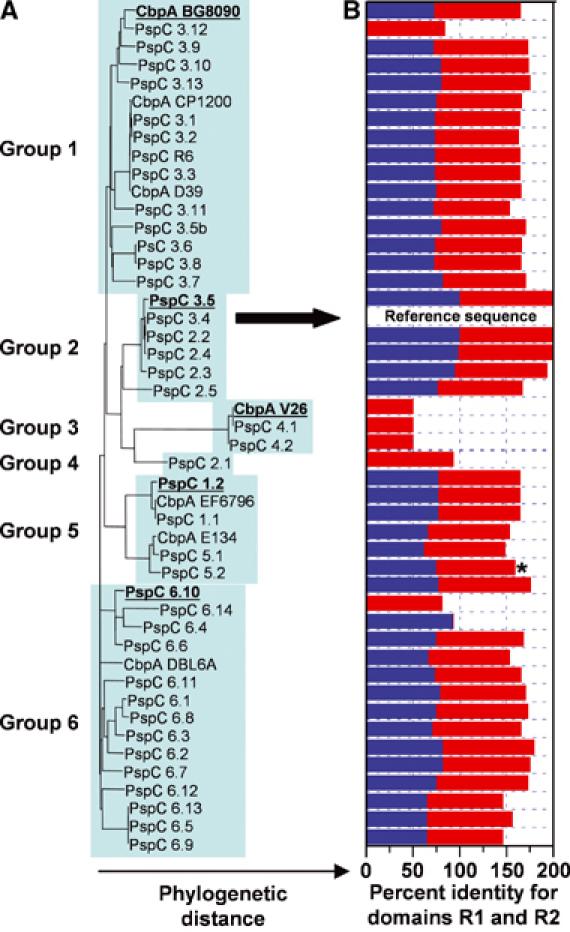
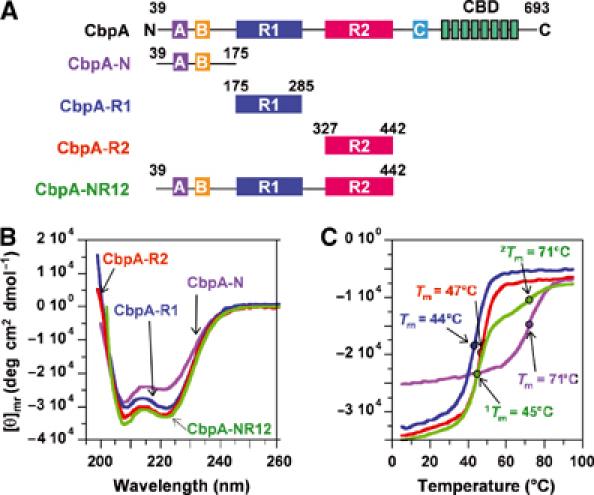
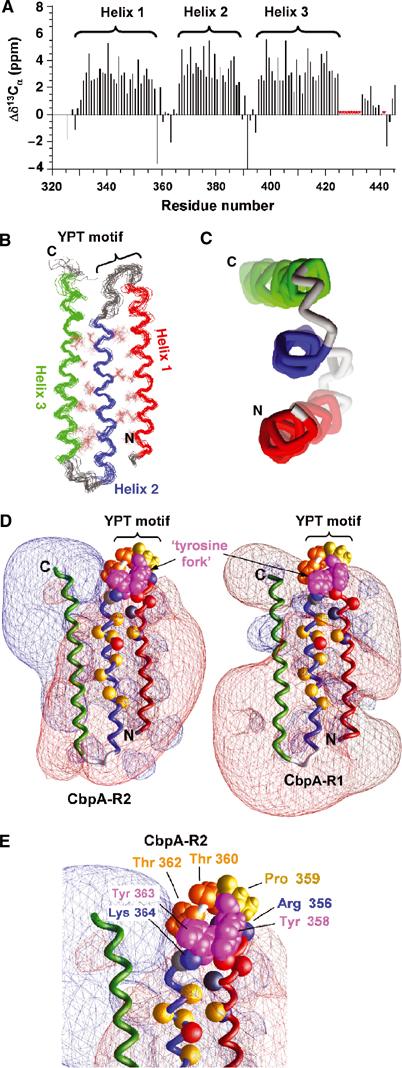
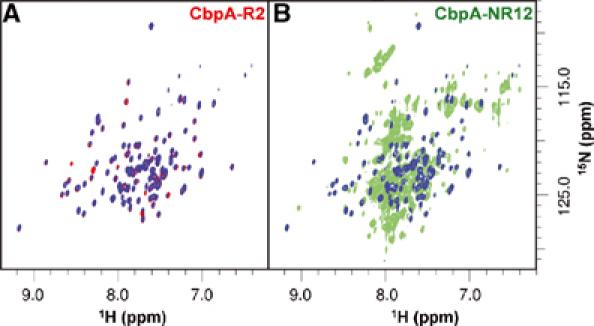
Streptococcus pneumoniae (pneumococcus) remains a significant health threat worldwide, especially to the young and old. While some of the biomolecules involved in pneumococcal pathogenesis are known and understood in mechanistic terms, little is known about the molecular details of bacterium/host interactions. We report here the solution structure of the 'repeated' adhesion domains (domains R1 and R2) of the principal pneumococcal adhesin, choline binding protein A (CbpA). Further, we provide insights into the mechanism by which CbpA binds its human receptor, polymeric immunoglobulin receptor (pIgR). The R domains, comprised of 12 imperfect copies of the leucine zipper heptad motif, adopt a unique 3-alpha-helix, raft-like structure. Each pair of alpha-helices is antiparallel and conserved residues in the loop between Helices 1 and 2 exhibit a novel 'tyrosine fork' structure that is involved in binding pIgR. This and other structural features that we show are conserved in most pneumococcal strains appear to generally play an important role in bacterial adhesion to pIgR. Interestingly, pneumococcus is the only bacterium known to adhere to and invade human cells by binding to pIgR.
Figures
References
-
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 - PubMed
-
- Butler JC, Breiman RF, Campbell JF, Lipman HB, Broome CV, Facklam RR (1993) Pneumococcal polysaccharide vaccine efficacy. An evaluation of current recommendations. JAMA 270: 1826–1831 - PubMed
-
- Case DA, Darden TA, Cheatham ITE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA (2004) AMBER 8. University of California, San Francisco, http://amber.scripps.edu/doc8/amber 8.pdf
-
- Cavanagh J, Fairbrother WJ, Palmer AG III, Skelton NJ (1996) Protein NMR Spectroscopy. New York: Academic Press
-
- Chemical_Computing_Group MOE version 2004.02. Montreal, Quebec, Canada
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
