

Phorbol 12-myristate 13-acetate induces epidermal growth factor receptor transactivation via protein kinase Cdelta/c-Src pathways in glioblastoma cells
- PMID: 15618223
- PMCID: PMC1351089
- DOI: 10.1074/jbc.M409056200
Phorbol 12-myristate 13-acetate induces epidermal growth factor receptor transactivation via protein kinase Cdelta/c-Src pathways in glioblastoma cells
Abstract
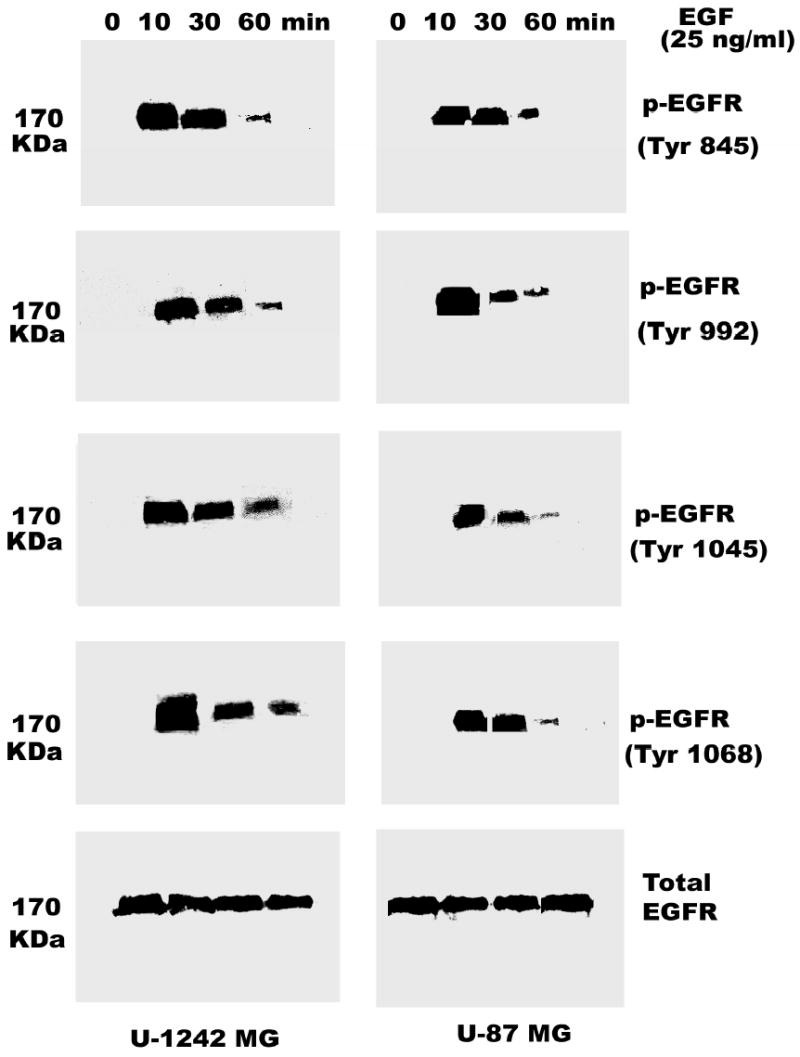
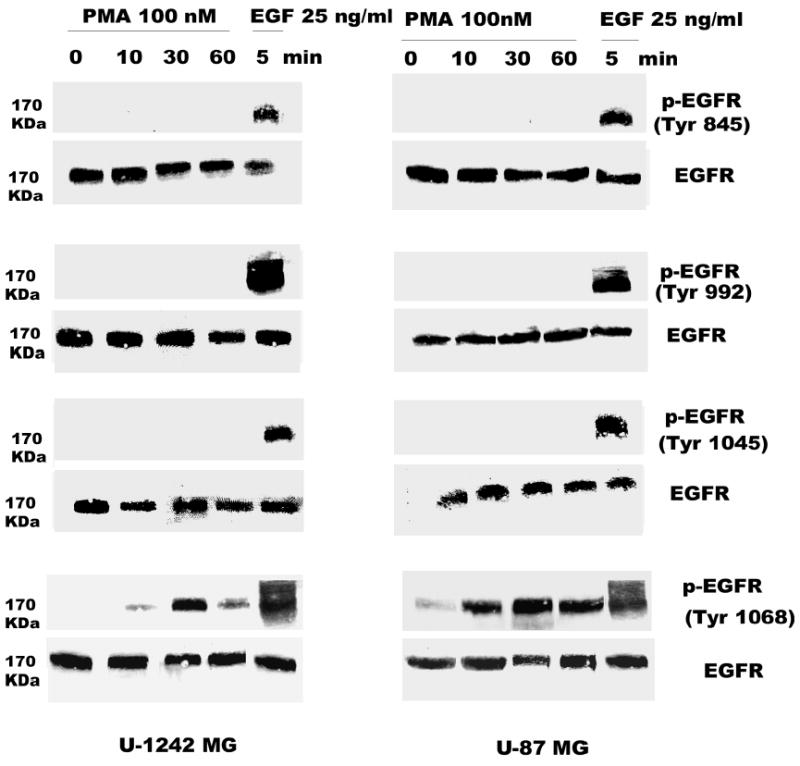
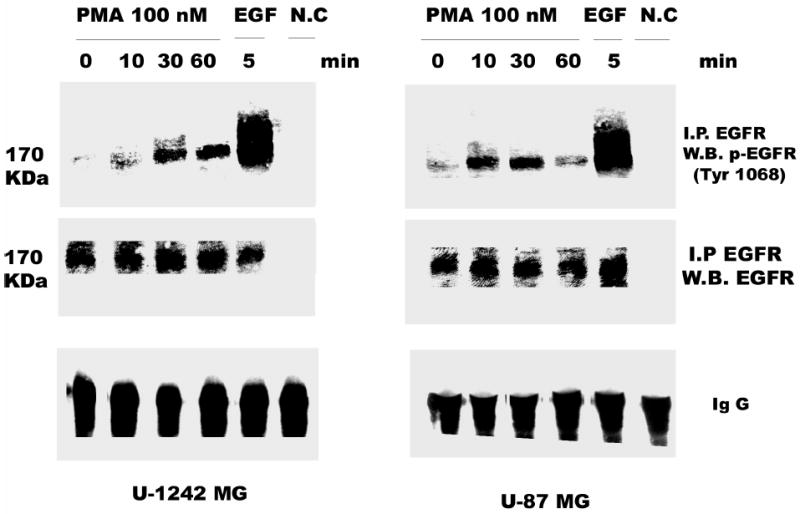
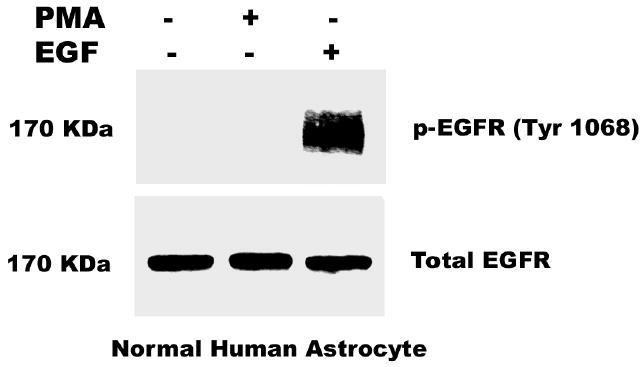
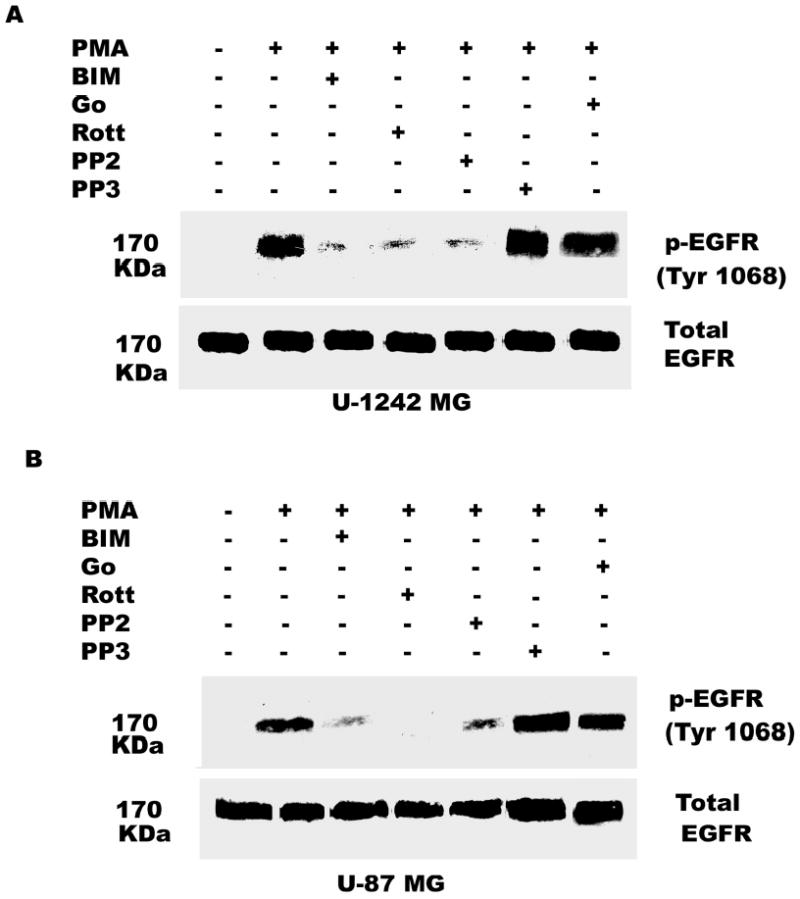
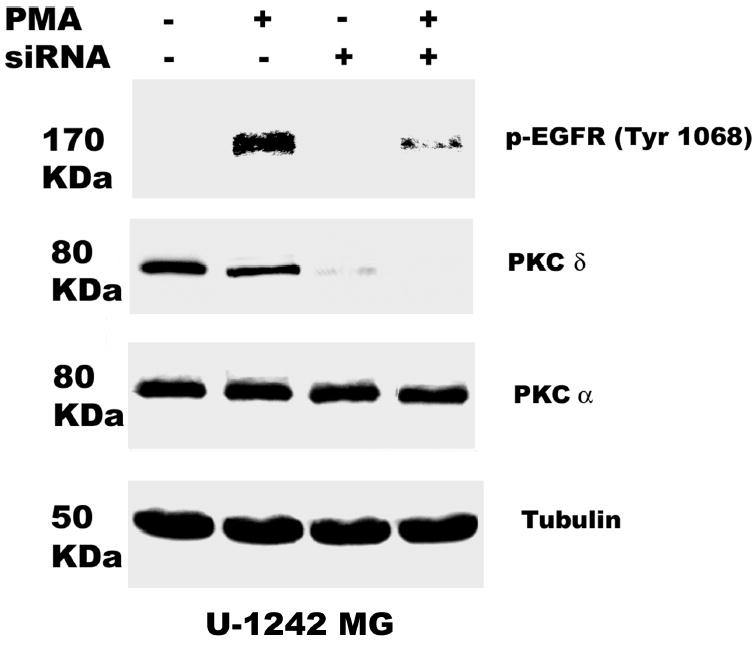
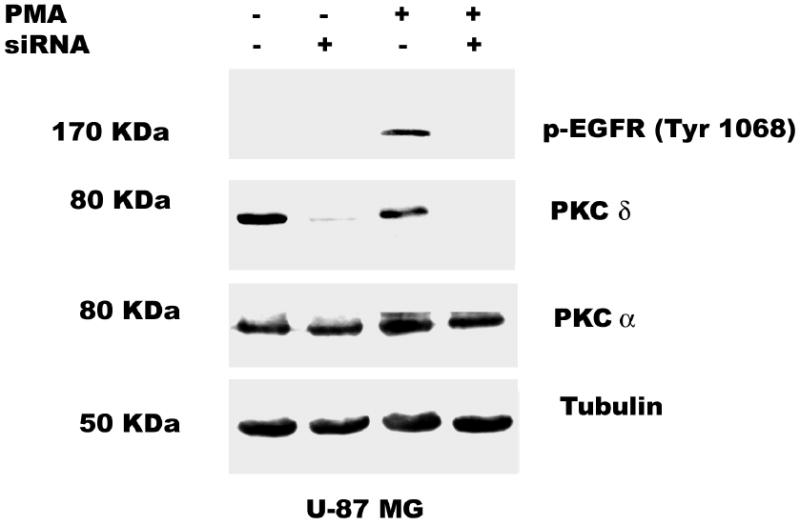
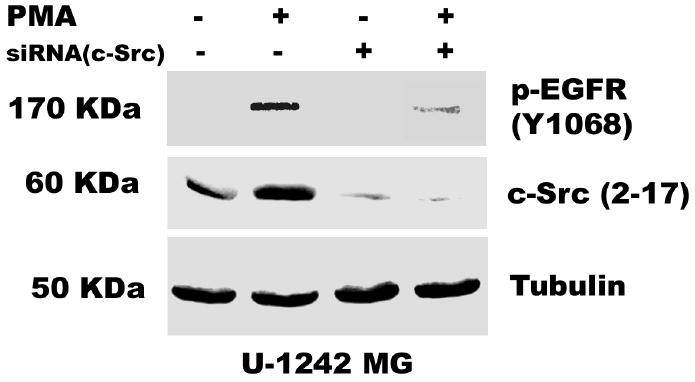
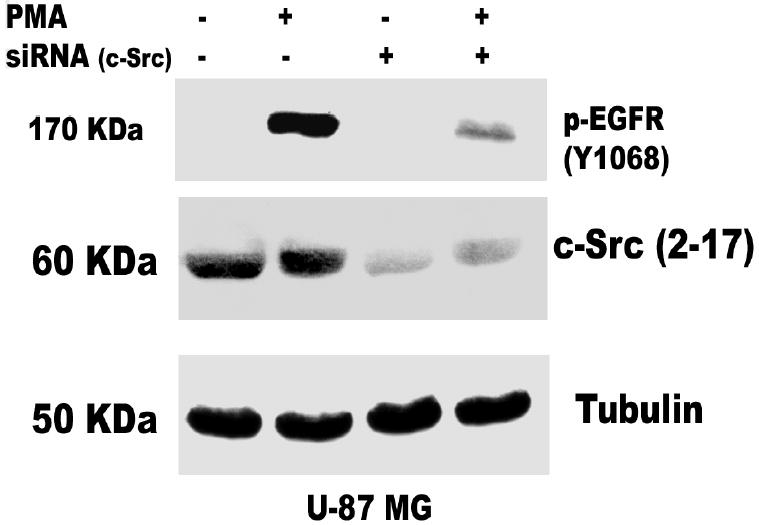
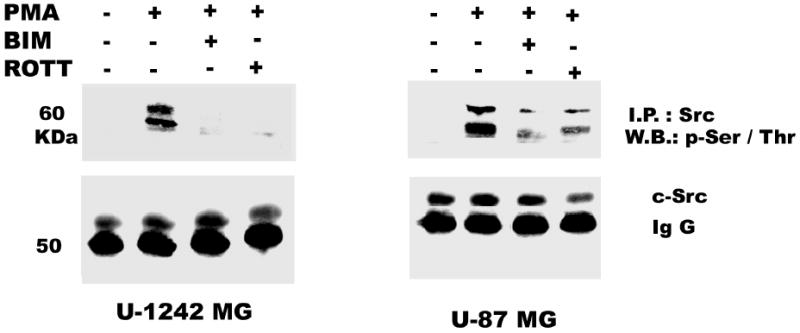
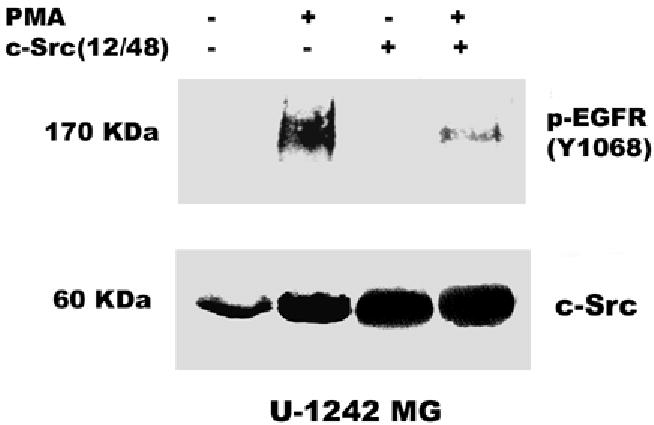
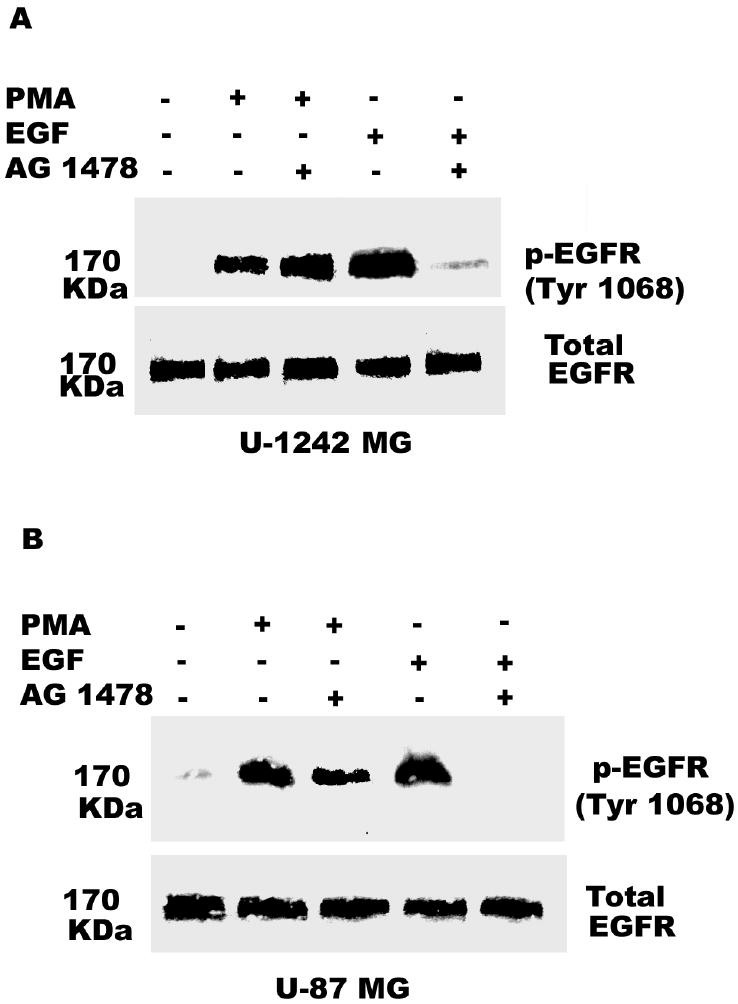
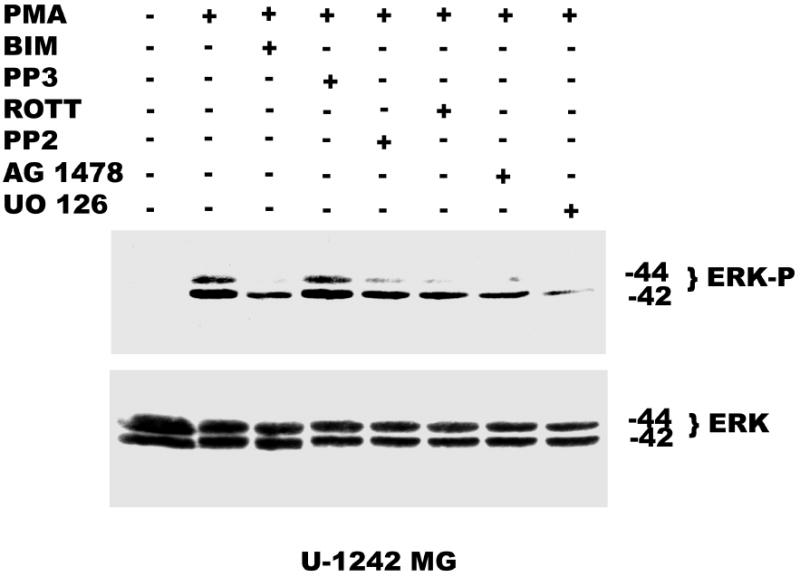
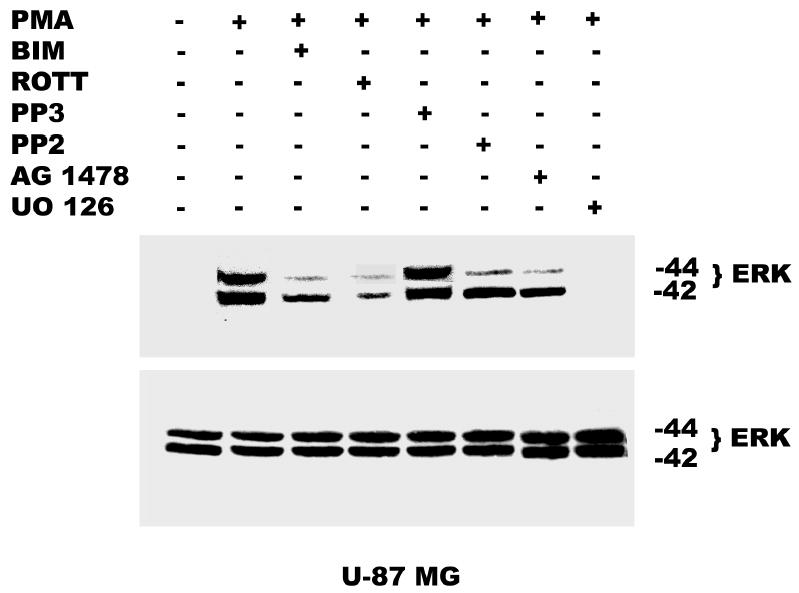
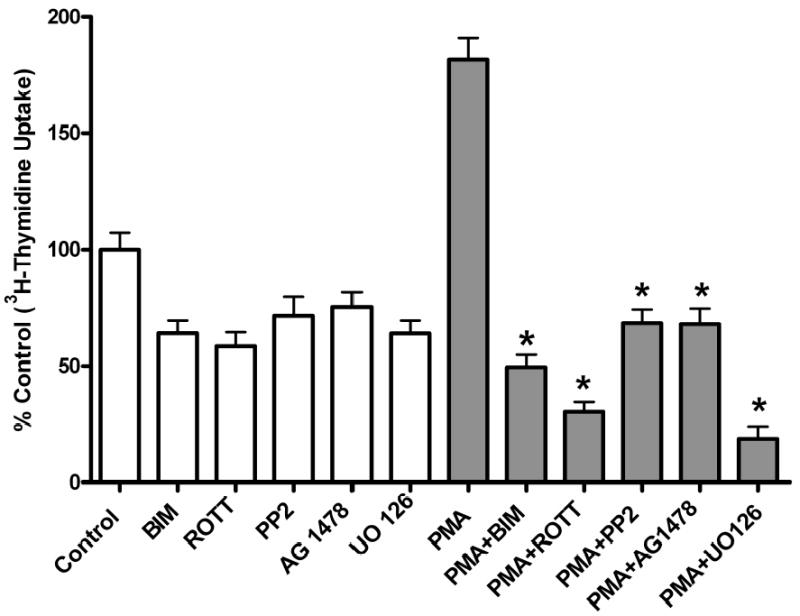
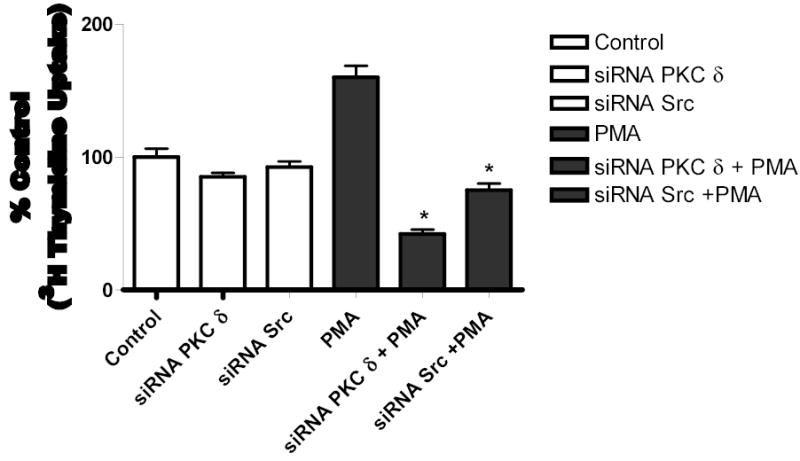
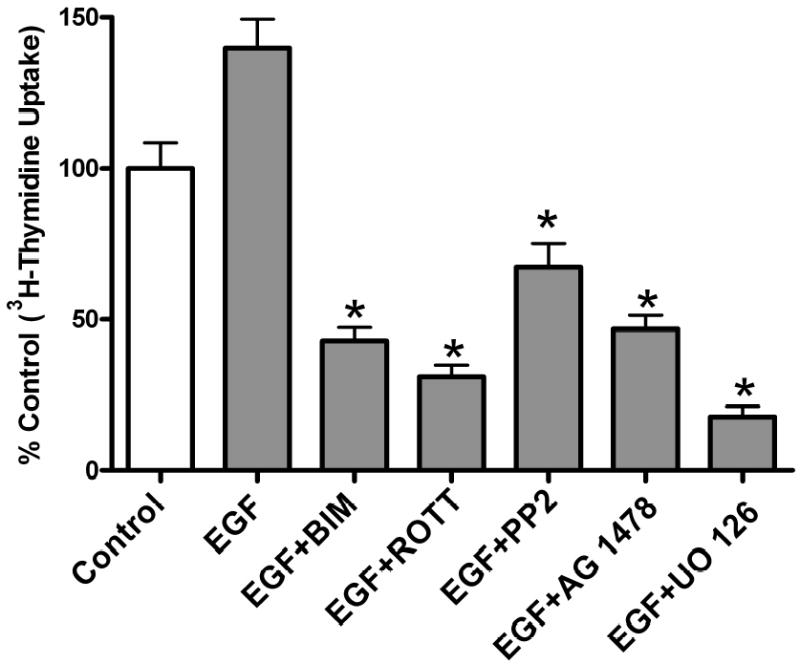
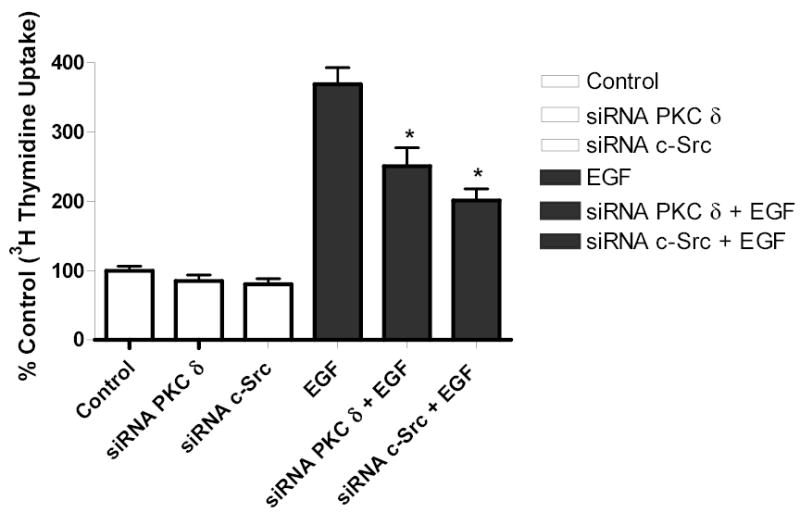
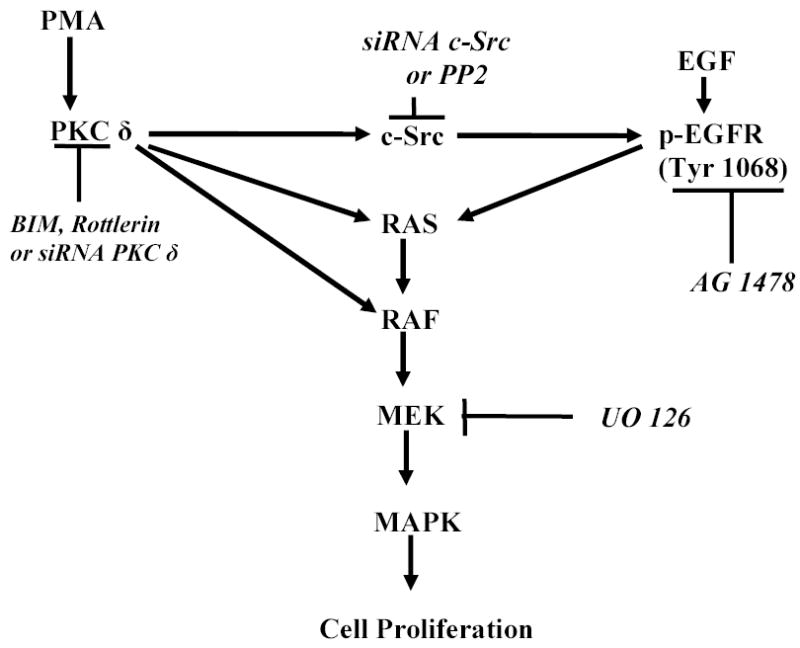
Both the epidermal growth factor receptor (EGFR) and protein kinase C (PKC) play important roles in glioblastoma invasive growth; however, the interaction between the EGFR and PKC is not well characterized in glioblastomas. Treatment with EGF stimulated global phosphorylation of the EGFR at Tyr(845), Tyr(992), Tyr(1068), and Tyr(1045) in glioblastoma cell lines (U-1242 MG and U-87 MG). Interestingly, phorbol 12-myristate 13-acetate (PMA) stimulated phosphorylation of the EGFR only at Tyr(1068) in the two glioblastoma cell lines. Phosphorylation of the EGFR at Tyr(1068) was not detected in normal human astrocytes treated with the phorbol ester. PMA-induced phosphorylation of the EGFR at Tyr(1068) was blocked by bisindolylmaleimide (BIM), a PKC inhibitor, and rottlerin, a PKCdelta-specific inhibitor. In contrast, Go 6976, an inhibitor of classical PKC isozymes, had no effect on PMA-induced EGFR phosphorylation. Furthermore, gene silencing with PKCdelta small interfering RNA (siRNA), siRNA against c-Src, and mutant c-Src(S12C/S48A) and treatment with a c-Src inhibitor (4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine) abrogated PMA-induced EGFR phosphorylation at Tyr(1068). PMA induced serine/threonine phosphorylation of Src, which was blocked by both BIM and rottlerin. Inhibition of the EGFR with AG 1478 did not significantly alter PMA-induced EGFR Tyr(1068) phosphorylation, but completely blocked EGF-induced phosphorylation of the EGFR. The effects of PMA on MAPK phosphorylation and glioblastoma cell proliferation were reduced by BIM, rottlerin, the MEK inhibitor U0126, and PKCdelta and c-Src siRNAs. Taken together, our data demonstrate that PMA transactivates the EGFR and increases cell proliferation by activating the PKCdelta/c-Src pathway in glioblastomas.
Figures
References
-
- Collins VP. Cancer Surv. 1998;32:37–51. - PubMed
-
- Rasheed BK, Wiltshire RN, Bigner SH, Bigner DD. Curr Opin Oncol. 1999;11:162–167. - PubMed
-
- Kleihues, P., Burger, P.C., Collins V.P., Newcomb E.W., Ohgaki, H., and Cavene, W.K., (2000). Glioblastoma. Tumors of the Nervous System. Kleihues, P., and Cavene, W.K. (eds) IARC Press: Lyon, pp 29–39.
-
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J. Nature. 1985;313:144–147. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
