

Transcription-based prediction of response to IFNbeta using supervised computational methods
- PMID: 15630474
- PMCID: PMC539058
- DOI: 10.1371/journal.pbio.0030002
Transcription-based prediction of response to IFNbeta using supervised computational methods
Abstract
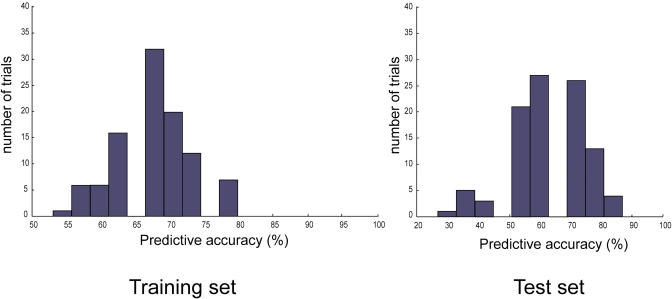
Changes in cellular functions in response to drug therapy are mediated by specific transcriptional profiles resulting from the induction or repression in the activity of a number of genes, thereby modifying the preexisting gene activity pattern of the drug-targeted cell(s). Recombinant human interferon beta (rIFNbeta) is routinely used to control exacerbations in multiple sclerosis patients with only partial success, mainly because of adverse effects and a relatively large proportion of nonresponders. We applied advanced data-mining and predictive modeling tools to a longitudinal 70-gene expression dataset generated by kinetic reverse-transcription PCR from 52 multiple sclerosis patients treated with rIFNbeta to discover higher-order predictive patterns associated with treatment outcome and to define the molecular footprint that rIFNbeta engraves on peripheral blood mononuclear cells. We identified nine sets of gene triplets whose expression, when tested before the initiation of therapy, can predict the response to interferon beta with up to 86% accuracy. In addition, time-series analysis revealed potential key players involved in a good or poor response to interferon beta. Statistical testing of a random outcome class and tolerance to noise was carried out to establish the robustness of the predictive models. Large-scale kinetic reverse-transcription PCR, coupled with advanced data-mining efforts, can effectively reveal preexisting and drug-induced gene expression signatures associated with therapeutic effects.
Figures






References
-
- Samuel CE, Knutson GS. Mechanism of interferon action: Human leukocyte and immune interferons regulate the expression of different genes and induce different antiviral states in human amnion U cells. Virology. 1983;130:474–484. - PubMed
-
- Tompkins WA. Immunomodulation and therapeutic effects of the oral use of interferon-alpha: Mechanism of action. J Interferon Cytokine Res. 1999;19:817–828. - PubMed
-
- Zhang X, Xu HT, Zhang CY, Liu JJ, Liu CM, et al. Immunomodulation of human cytomegalovirus infection on interferon system in patients with systemic lupus erythematosus. J Tongji Med Univ. 1991;11:126–128. - PubMed
-
- Arnason BG. Interferon beta in multiple sclerosis. Clin Immunol Immunopathol. 1996;81:1–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
