

Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors
- PMID: 15647351
- PMCID: PMC545566
- DOI: 10.1073/pnas.0409143102
Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors
Abstract
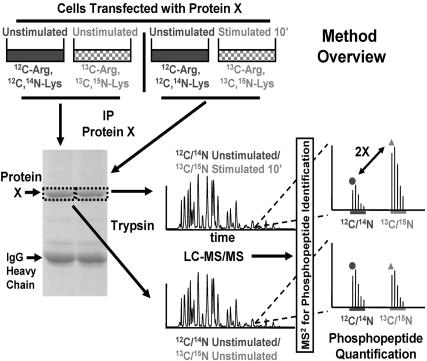
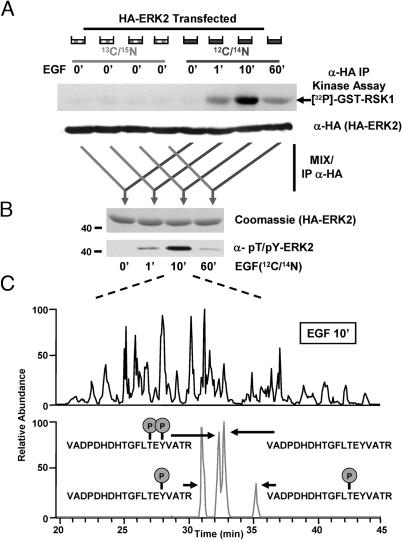
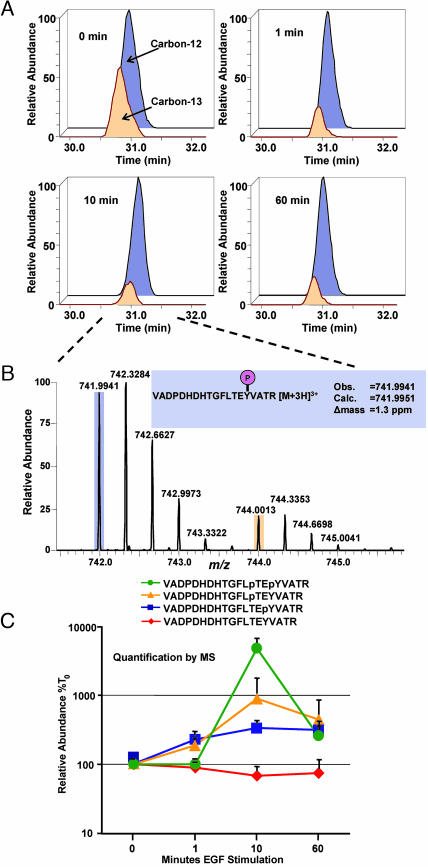
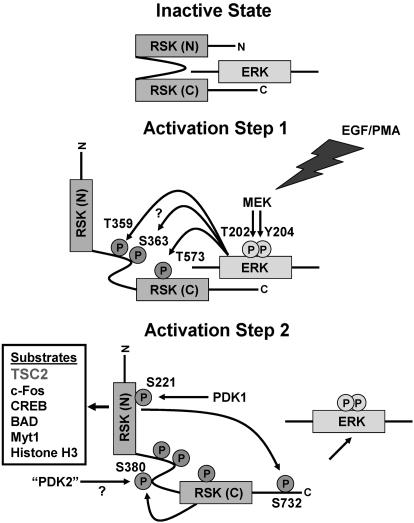
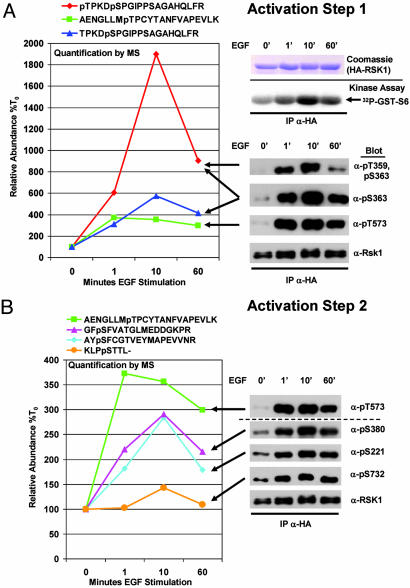
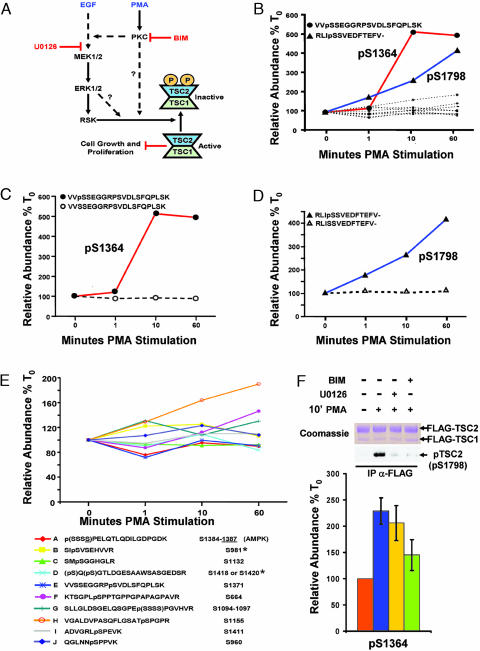
Reversible protein phosphorylation is an essential cellular regulatory mechanism. Many proteins integrate and are modulated by multiple phosphorylation events derived from complex signaling cues. Simultaneous detection and quantification of temporal changes in all of a protein's phosphorylation sites could provide not only an immediate assessment of a known biochemical activity but also important insights into molecular signaling mechanisms. Here we show the use of stable isotope-based quantitative MS to globally monitor the kinetics of complex, ordered phosphorylation events on protein players in the canonical mitogen-activated protein kinase signaling pathway. In excellent agreement with activity assays and phosphospecific immunoblotting with the same samples, we quantified epidermal growth factor-induced changes in nine phosphorylation sites in the extracellular signal-regulated kinase (ERK)/p90 ribosomal S6 kinase-signaling cassette. Additionally, we monitored 14 previously uncharacterized and six known phosphorylation events after phorbol ester stimulation in the ERK/p90 ribosomal S6 kinase-signaling targets, the tuberous sclerosis complex (TSC) tumor suppressors TSC1 and TSC2. By using quantitative phosphorylation profiling in conjunction with pharmacological kinase inhibitors we uncovered a ERK-independent, protein kinase C-dependent pathway to TSC2 phosphorylation. These results establish quantitative phosphorylation profiling as a means to simultaneously identify, quantify, and delineate the kinetic changes of ordered phosphorylation events on a given protein and defines parameters for the rapid discovery of important in vivo phosphoregulatory mechanisms.
Figures
References
-
- Ong, S. E., Blagoev, B., Kratchmarova, I., Kristensen, D. B., Steen, H., Pandey, A. & Mann, M. (2002) Mol. Cell Proteomics 1, 376-386. - PubMed
-
- Chen, X., Smith, L. M. & Bradbury, E. M. (2000) Anal. Chem. 72, 1134-1143. - PubMed
-
- Blagoev, B., Ong, S. E., Kratchmarova, I. & Mann, M. (2004) Nat. Biotechnol. 22, 1139-1145. - PubMed
-
- Ibarrola, N., Kalume, D. E., Gronborg, M., Iwahori, A. & Pandey, A. (2003) Anal. Chem. 75, 6043-6049. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
