

Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication
- PMID: 15650183
- PMCID: PMC544105
- DOI: 10.1128/JVI.79.3.1569-1580.2005
Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication
Retraction in
-
Retraction for Waris et al., "Hepatitis C Virus (HCV) Constitutively Activates STAT-3 via Oxidative Stress: Role of STAT-3 in HCV Replication".J Virol. 2020 Sep 15;94(19):e01358-20. doi: 10.1128/JVI.01358-20. Print 2020 Sep 15. J Virol. 2020. PMID: 32934069 Free PMC article. No abstract available.
Abstract
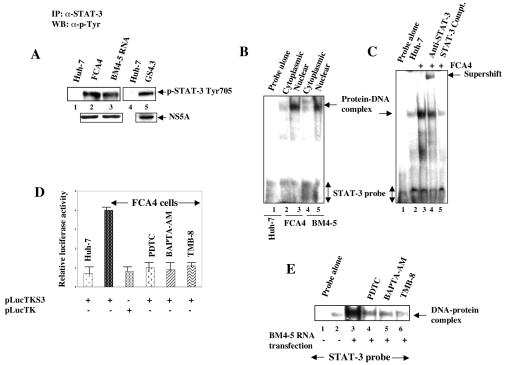
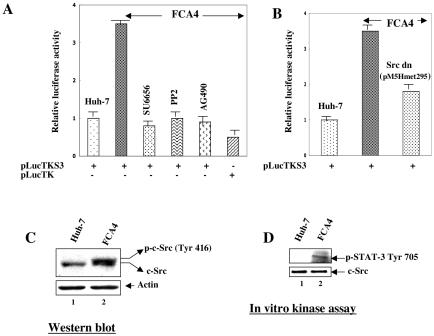
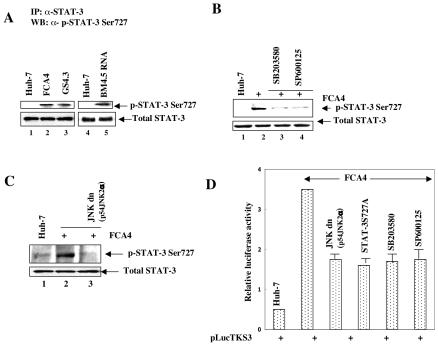
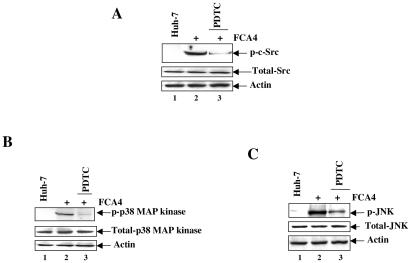
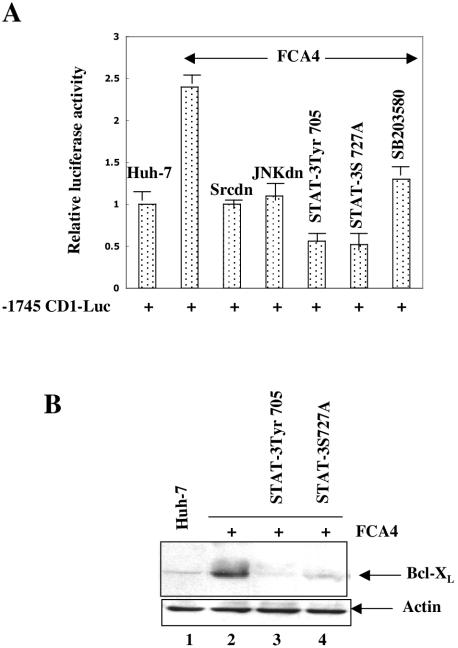
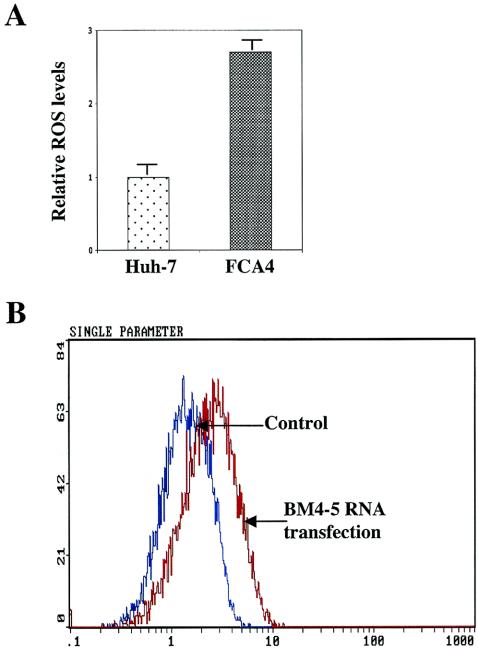
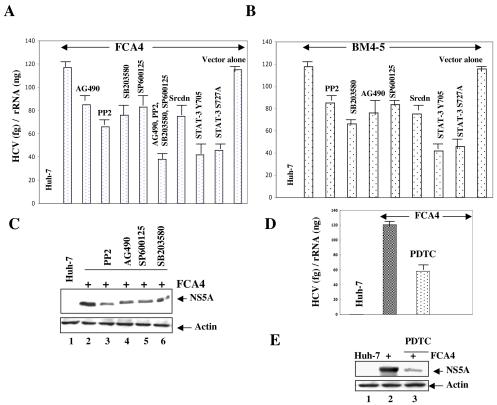
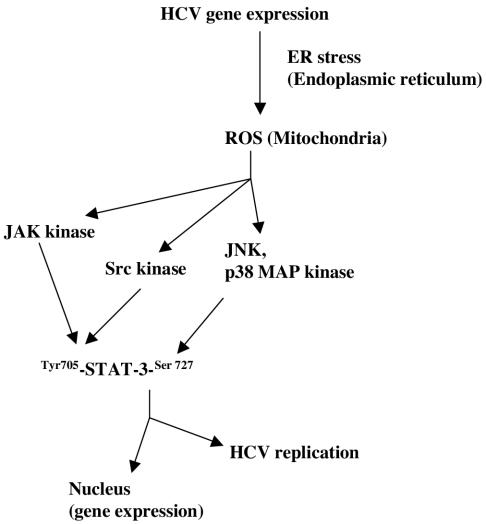
The hepatitis C virus (HCV) causes chronic hepatitis, which often results in liver cirrhosis and hepatocellular carcinoma. We have previously shown that HCV nonstructural proteins induce activation of STAT-3 via oxidative stress and Ca2+ signaling (G. Gong, G. Waris, R. Tanveer, and A. Siddiqui, Proc. Natl. Acad. Sci. USA 98:9599-9604, 2001). In this study, we focus on the signaling pathway leading to STAT-3 activation in response to oxidative stress induced by HCV translation and replication activities. Here, we demonstrate the constitutive activation of STAT-3 in HCV replicon-expressing cells. The HCV-induced STAT-3 activation was inhibited in the presence of antioxidant (pyrrolidine dithiocarbamate) and Ca2+ chelators (BAPTA-AM and TMB-8). Previous studies have shown that maximum STAT-3 transactivation requires Ser727 phosphorylation in addition to tyrosine phosphorylation. Using a series of inhibitors and dominant negative mutants, we show that HCV-induced activation of STAT-3 is mediated by oxidative stress and influenced by the activation of cellular kinases, including p38 mitogen-activated protein kinase, JNK, JAK-2, and Src. Our results also suggest a potential role of STAT-3 in HCV RNA replication. We also observed the constitutive activation of STAT-3 in the liver biopsy of an HCV-infected patient. These studies provide an insight into the mechanisms by which HCV induces intracellular events relevant to liver pathogenesis associated with the viral infection.
Figures


References
-
- Abe, J., M. Takahashi, M. Ishida, J. W. Lee, and B. C. Berk. 1997. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1 (BMK1). J. Biol. Chem. 272:20389-20394. - PubMed
-
- Bartenschlager, R., and V. Lohmann. 2000. Replication of hepatitis C virus. J. Gen. Virol. 81:1631-1648. - PubMed
-
- Bartenschlager, R., and V. Lohmann. 2001. Novel cell culture systems for the hepatitis C virus. Antivir. Res. 52:1-17. - PubMed
-
- Bowman, T., R. Garcia, J. Turkson, and R. Jove. 2000. STATs in oncogenesis. Oncogene 19:2474-2488. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
