

Combining data from multiple inbred line crosses improves the power and resolution of quantitative trait loci mapping
- PMID: 15654110
- PMCID: PMC1449540
- DOI: 10.1534/genetics.104.033993
Combining data from multiple inbred line crosses improves the power and resolution of quantitative trait loci mapping
Abstract
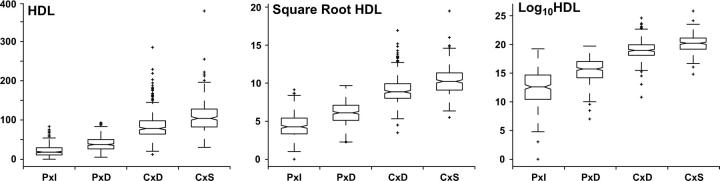
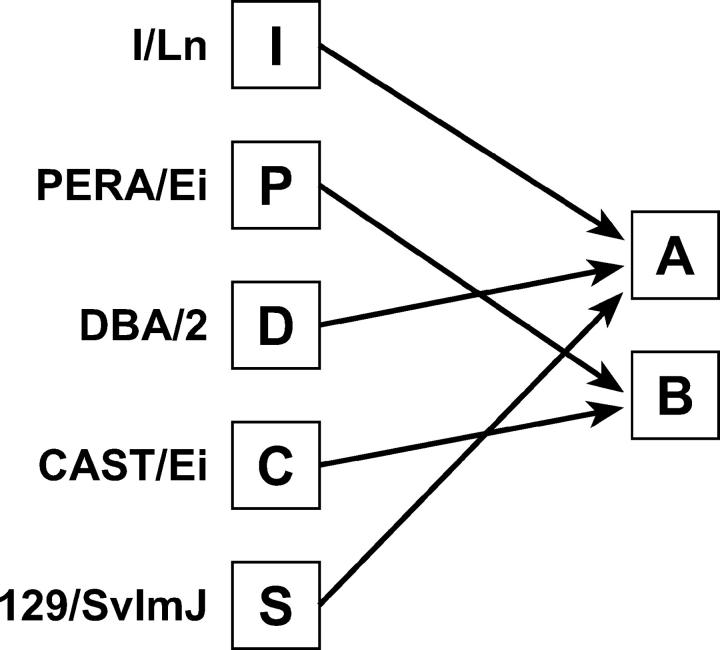
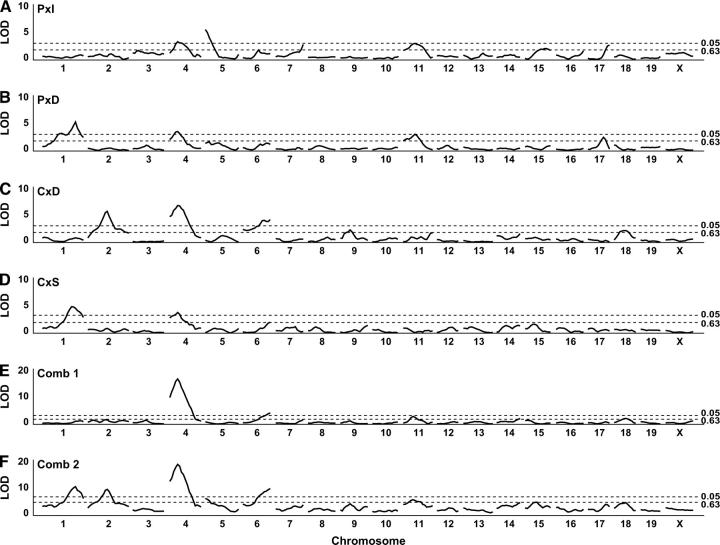
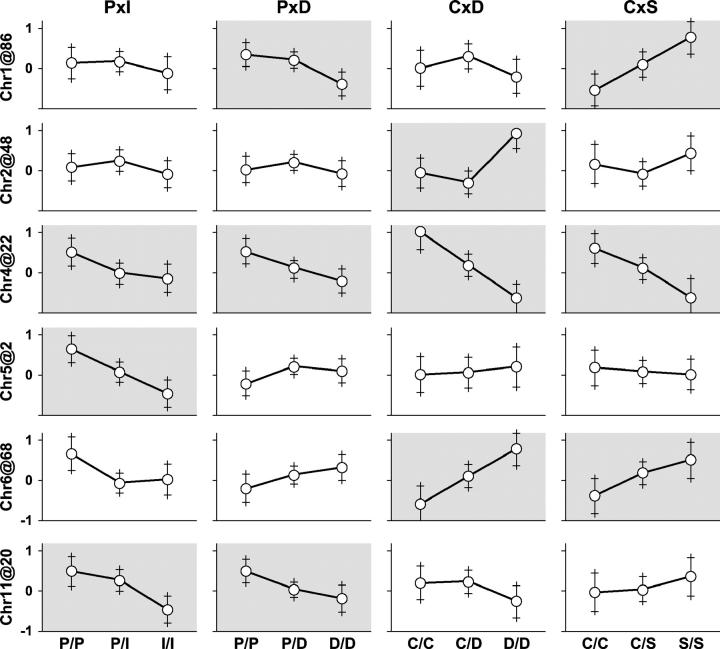
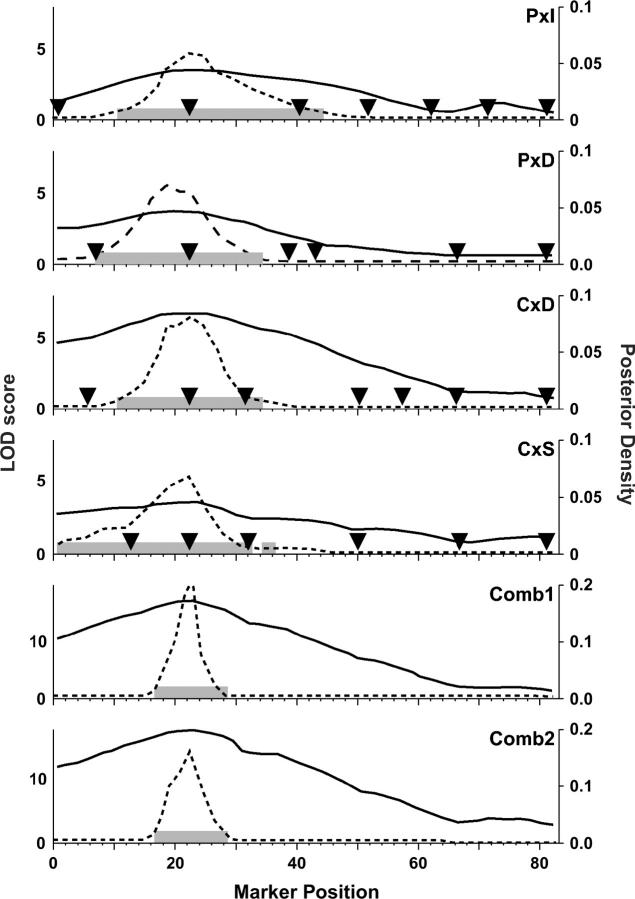
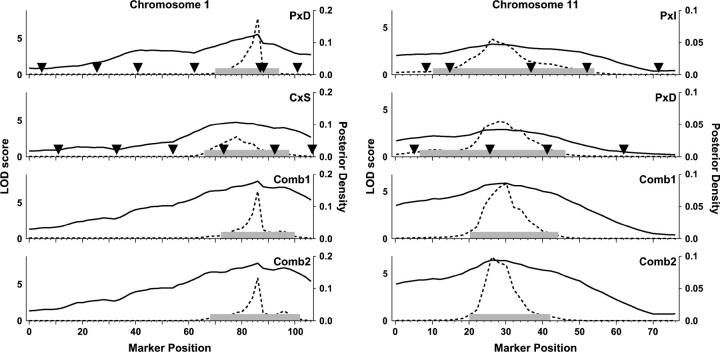
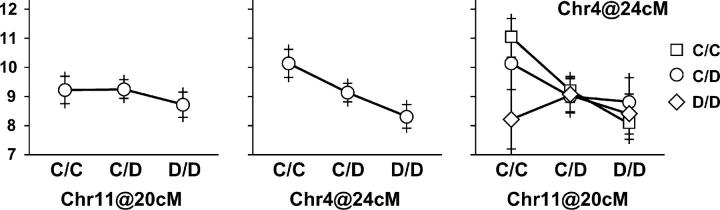
Rodent inbred line crosses are widely used to map genetic loci associated with complex traits. This approach has proven to be powerful for detecting quantitative trait loci (QTL); however, the resolution of QTL locations, typically approximately 20 cM, means that hundreds of genes are implicated as potential candidates. We describe analytical methods based on linear models to combine information available in two or more inbred line crosses. Our strategy is motivated by the hypothesis that common inbred strains of the laboratory mouse are derived from a limited ancestral gene pool and thus QTL detected in multiple crosses are likely to represent shared ancestral polymorphisms. We demonstrate that the combined-cross analysis can improve the power to detect weak QTL, can narrow support intervals for QTL regions, and can be used to separate multiple QTL that colocalize by chance. Moreover, combined-cross analysis can establish the allelic states of a QTL among a set of parental lines, thus providing critical information for narrowing QTL regions by haplotype analysis.
Figures
References
-
- Beck, J. A., S. Lloyd, M. Hafezparast, M. Lennon-Pierce, J. T. Eppig et al., 2000. Genealogies of mouse inbred strains. Nat. Genet. 24: 23–25. - PubMed
-
- Bonhomme, F., 1986. Evolutionary relationships in the genus Mus. Curr. Top. Microbiol. Immunol. 127: 19–34. - PubMed
-
- Broman, K. W., H. Wu, S. Sen and G. A. Churchill, 2003. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
