

Phenotypic perturbation of B cells in the Wiskott-Aldrich syndrome
- PMID: 15654828
- PMCID: PMC1809280
- DOI: 10.1111/j.1365-2249.2005.02693.x
Phenotypic perturbation of B cells in the Wiskott-Aldrich syndrome
Erratum in
- Clin Exp Immunol. 2005 May;140(2):394
Abstract
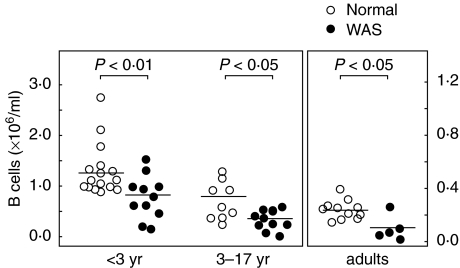
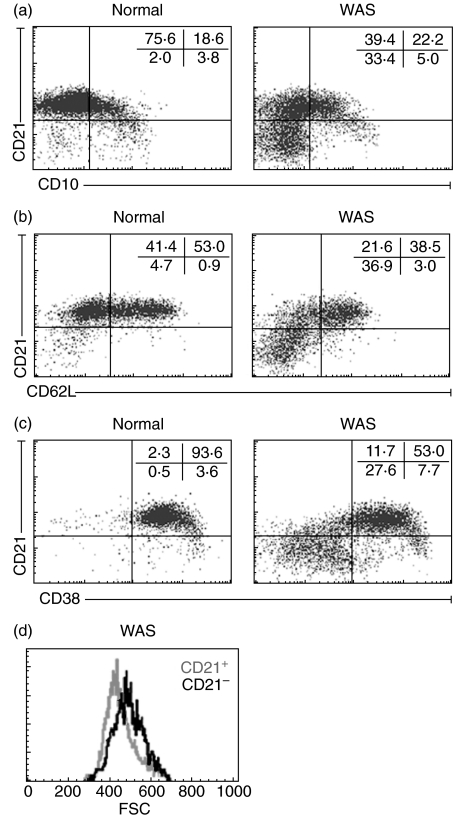
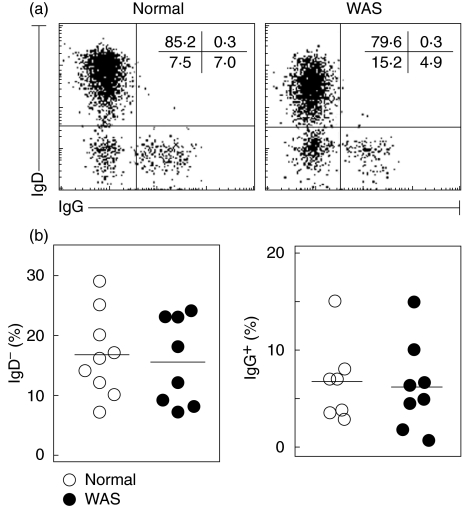
Wiskott-Aldrich syndrome (WAS) is an X-linked immunodeficiency/platelet disease due to mutations of WASP, a cytoskeletal regulatory protein of blood cells. Patients exhibit a range of immune defects generally attributed to defective T-cell function, including poor response to immunization, skewed immunoglobulin isotypes, eczema, recurrent infections, autoimmune disease and increased frequency of malignancies. Here we show a deficit of total B-cells in WAS patients of various ages and identify phenotypic perturbations involving complement receptors and CD27. Whereas B-cells of normal healthy donors are overwhelmingly CD21/CD35-positive, B-cells expressing these receptors are significantly reduced in number in WAS patients, and their paucity may cause suboptimal antigen capture and presentation. The frequencies of IgD(-) and IgG(+) patient B-cells were not different from healthy donors (although absolute numbers were decreased), indicating that isotype switching is occurring. In contrast, the frequency of cells positive for CD27, the marker of post germinal centre B-cells, was significantly decreased even among isotype-switched cells, and B-cells resembling germinal centre progenitors (CD10(+)CD27(-)CD38(bright)) were more frequent in adult patients, suggesting impaired germinal centre maturation/differentiation. The documentation of these phenotypic perturbations and deficit of total cells suggest that defects intrinsic to B-cells contribute to the impaired humoral immunity that characterizes this disease.
Figures



Similar articles
-
Wiskott-Aldrich Syndrome protein deficiency perturbs the homeostasis of B-cell compartment in humans.J Autoimmun. 2014 May;50(100):42-50. doi: 10.1016/j.jaut.2013.10.006. Epub 2013 Dec 25. J Autoimmun. 2014. PMID: 24369837 Free PMC article.
-
Early deficit of lymphocytes in Wiskott-Aldrich syndrome: possible role of WASP in human lymphocyte maturation.Clin Exp Immunol. 2004 Apr;136(1):104-10. doi: 10.1111/j.1365-2249.2004.02409.x. Clin Exp Immunol. 2004. PMID: 15030520 Free PMC article.
-
Effects of Wiskott-Aldrich Syndrome Protein Deficiency on IL-10-Producing Regulatory B Cells in Humans and Mice.Scand J Immunol. 2015 Jun;81(6):483-93. doi: 10.1111/sji.12282. Scand J Immunol. 2015. PMID: 25728049
-
The Wiskott-Aldrich syndrome protein (WASP): roles in signaling and cytoskeletal organization.Annu Rev Immunol. 1999;17:905-29. doi: 10.1146/annurev.immunol.17.1.905. Annu Rev Immunol. 1999. PMID: 10358777 Review.
-
The Wiskott-Aldrich syndrome.Semin Hematol. 1998 Oct;35(4):332-45. Semin Hematol. 1998. PMID: 9801262 Review.
Cited by
-
Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency.Front Immunol. 2012 Aug 27;3:265. doi: 10.3389/fimmu.2012.00265. eCollection 2012. Front Immunol. 2012. PMID: 22969765 Free PMC article.
-
Common Variable Immunodeficiency and Autoimmune Diseases: A Retrospective Study of 95 Adult Patients in a Single Tertiary Care Center.Front Immunol. 2021 Jul 5;12:652487. doi: 10.3389/fimmu.2021.652487. eCollection 2021. Front Immunol. 2021. PMID: 34290696 Free PMC article.
-
Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott-Aldrich Syndrome.J Clin Immunol. 2018 Jan;38(1):13-27. doi: 10.1007/s10875-017-0453-z. Epub 2017 Oct 30. J Clin Immunol. 2018. PMID: 29086100 Review.
-
Immature B cells preferentially switch to IgE with increased direct Sμ to Sε recombination.J Exp Med. 2011 Dec 19;208(13):2733-46. doi: 10.1084/jem.20111155. Epub 2011 Dec 5. J Exp Med. 2011. PMID: 22143888 Free PMC article.
-
Actin cytoskeletal defects in immunodeficiency.Immunol Rev. 2013 Nov;256(1):282-99. doi: 10.1111/imr.12114. Immunol Rev. 2013. PMID: 24117828 Free PMC article. Review.
References
-
- Wiskott A. Familiarer, angeobren Morbus Werlhofi? Monatschrift Kinderheil. 1937;68:212–6.
-
- Aldrich R, Steinberg A, Campbell D. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics. 1954;13:133–8. - PubMed
-
- Sullivan KE. Recent advances in our understanding of Wiskott–Aldrich syndrome. Curr Opin Hematol. 1999;6:8–14. - PubMed
-
- Remold-O’Donnell E, Rosen FS, Kenney DM. Defects in Wiskott–Aldrich syndrome blood cells. Blood. 1996;87:2621–31. - PubMed
-
- Ochs HD, Rosen FS. Primary Immunodeficiency Diseases. New York: Oxford University Press; 1999.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
