

Inhibition of macroautophagy triggers apoptosis
- PMID: 15657430
- PMCID: PMC543994
- DOI: 10.1128/MCB.25.3.1025-1040.2005
Inhibition of macroautophagy triggers apoptosis
Abstract
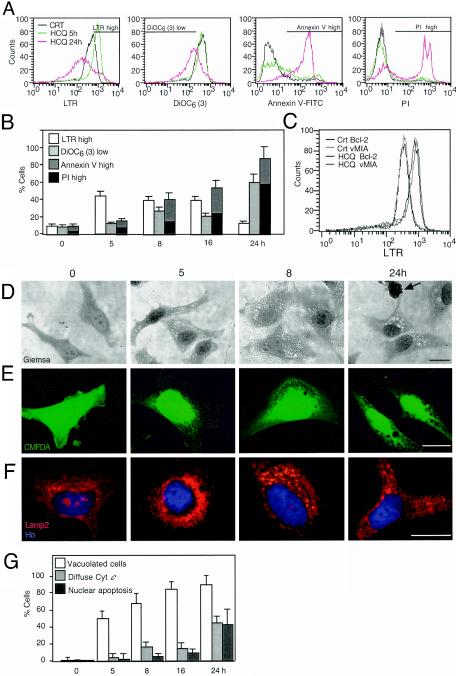
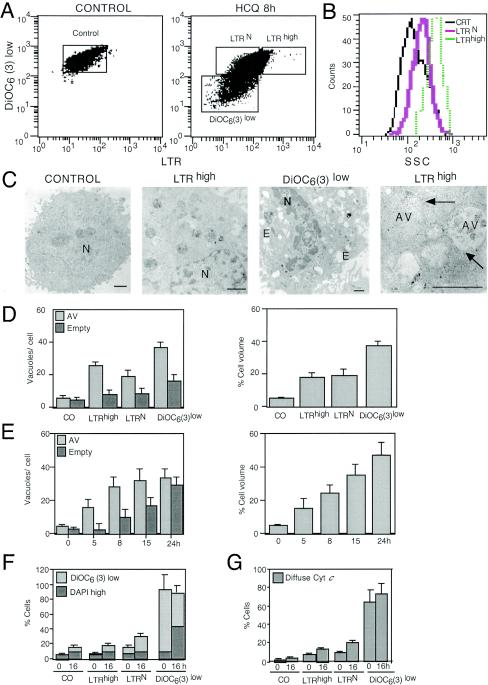
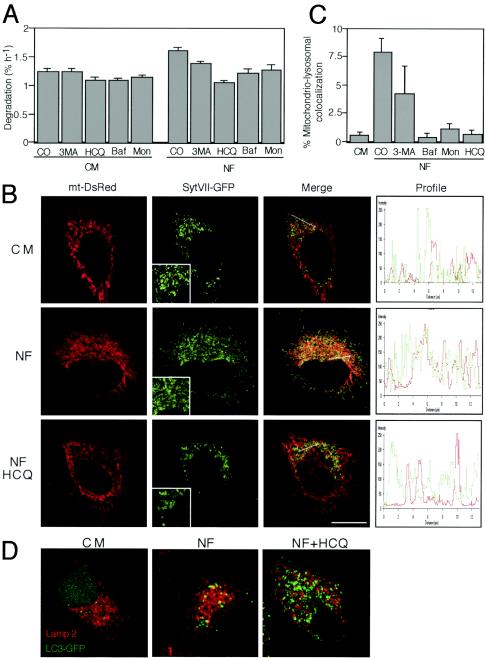
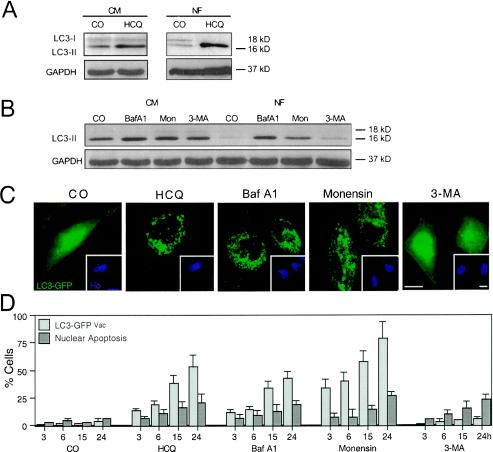
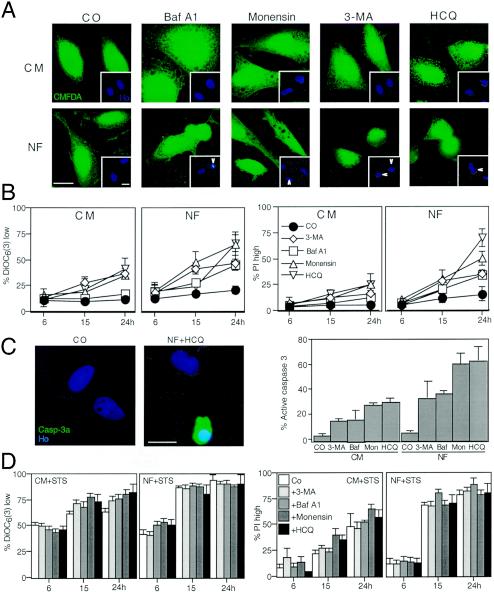
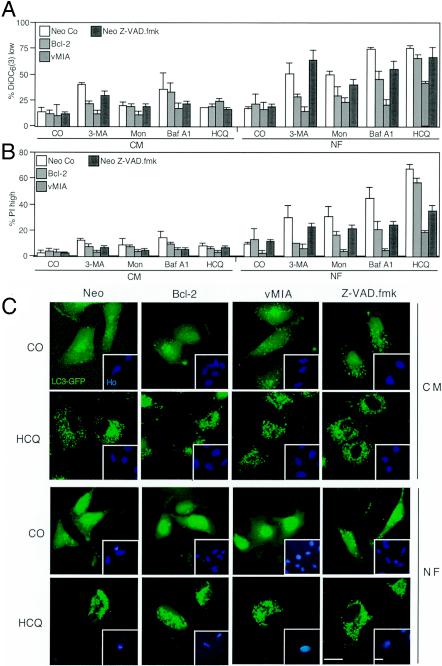
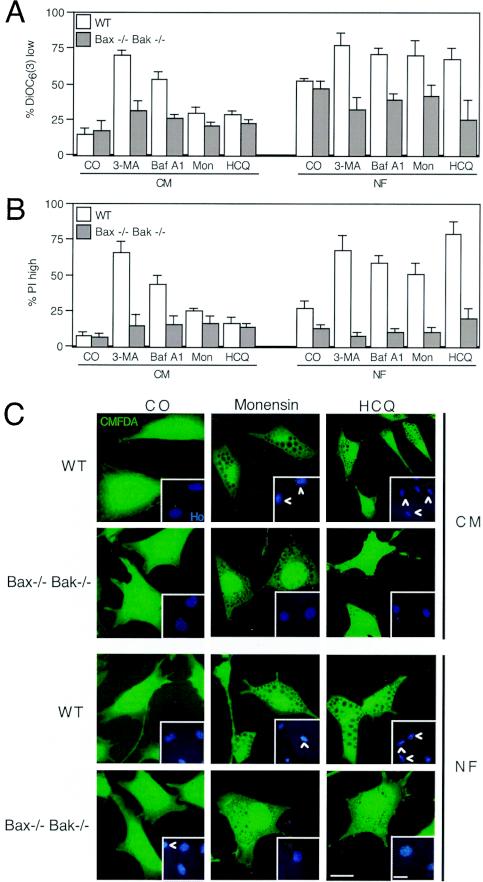
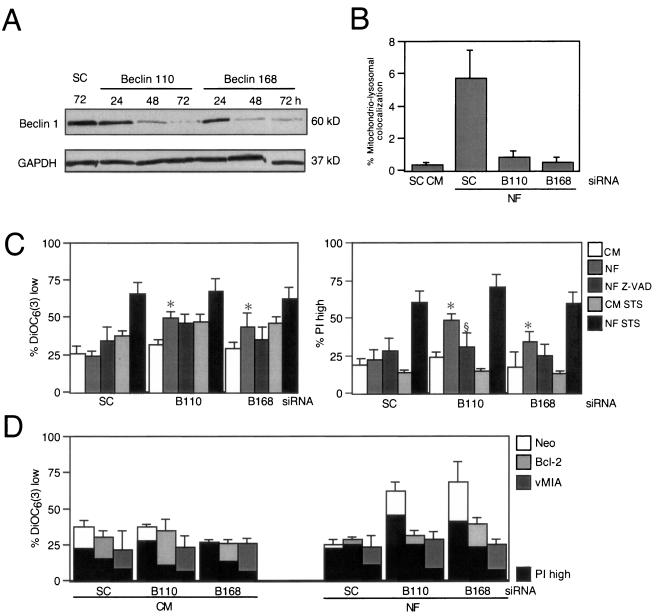
Mammalian cells were observed to die under conditions in which nutrients were depleted and, simultaneously, macroautophagy was inhibited either genetically (by a small interfering RNA targeting Atg5, Atg6/Beclin 1-1, Atg10, or Atg12) or pharmacologically (by 3-methyladenine, hydroxychloroquine, bafilomycin A1, or monensin). Cell death occurred through apoptosis (type 1 cell death), since it was reduced by stabilization of mitochondrial membranes (with Bcl-2 or vMIA, a cytomegalovirus-derived gene) or by caspase inhibition. Under conditions in which the fusion between lysosomes and autophagosomes was inhibited, the formation of autophagic vacuoles was enhanced at a preapoptotic stage, as indicated by accumulation of LC3-II protein, ultrastructural studies, and an increase in the acidic vacuolar compartment. Cells exhibiting a morphology reminiscent of (autophagic) type 2 cell death, however, recovered, and only cells with a disrupted mitochondrial transmembrane potential were beyond the point of no return and inexorably died even under optimal culture conditions. All together, these data indicate that autophagy may be cytoprotective, at least under conditions of nutrient depletion, and point to an important cross talk between type 1 and type 2 cell death pathways.
Figures
References
-
- Adams, J. M. 2003. Ways of dying: multiple pathways to apoptosis. Genes Dev. 17:2481-2495. - PubMed
-
- Beers, M. F. 1996. Inhibition of cellular processing of surfactant protein C by drugs affecting intracellular pH gradients. J. Biol. Chem. 271:14361-14370. - PubMed
-
- Belzacq, A. S., C. El Hamel, H. L. A. Vieira, I. Cohen, D. Haouzi, D. Métivier, P. Marchetti, V. Goldmacher, C. Brenner, and G. Kroemer. 2001. The adenine nucleotide translocator mediates the mitochondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene 20:7579-7587. - PubMed
-
- Blommaart, E. F., U. Krause, J. P. Schellens, H. Vreeling-Sindelarova, and A. J. Meijer. 1997. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 243:240-246. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases