

Using large-scale perturbations in gene network reconstruction
- PMID: 15659246
- PMCID: PMC548128
- DOI: 10.1186/1471-2105-6-11
Using large-scale perturbations in gene network reconstruction
Abstract
Background: Recent analysis of the yeast gene network shows that most genes have few inputs, indicating that enumerative gene reconstruction methods are both useful and computationally feasible. A simple enumerative reconstruction method based on a discrete dynamical system model is used to study how microarray experiments involving modulated global perturbations can be designed to obtain reasonably accurate reconstructions. The method is tested on artificial gene networks with biologically realistic in/out degree characteristics.
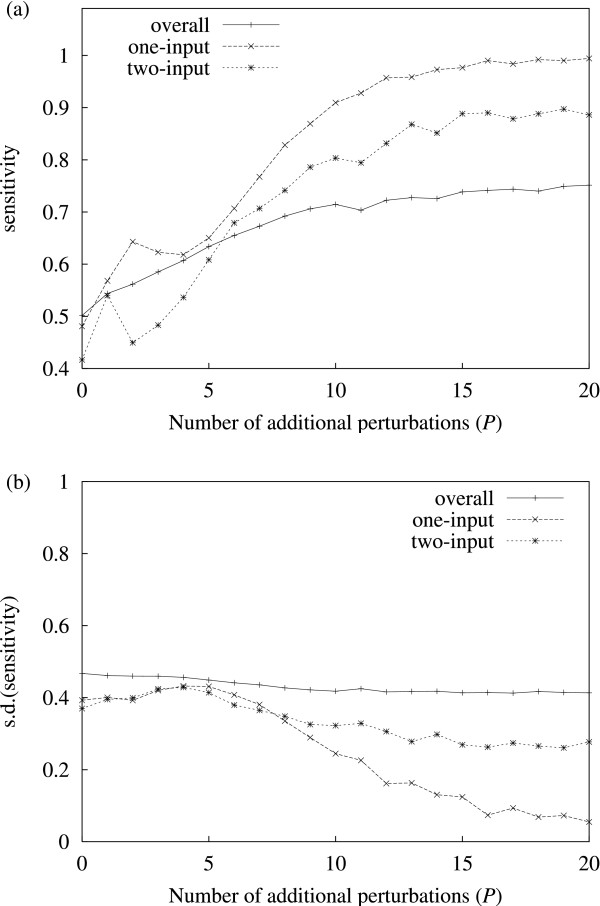
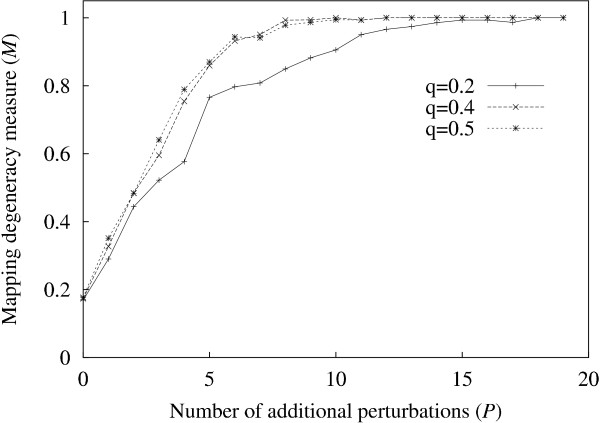
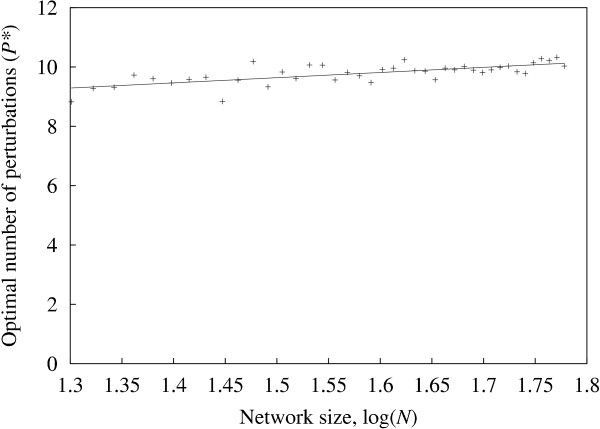
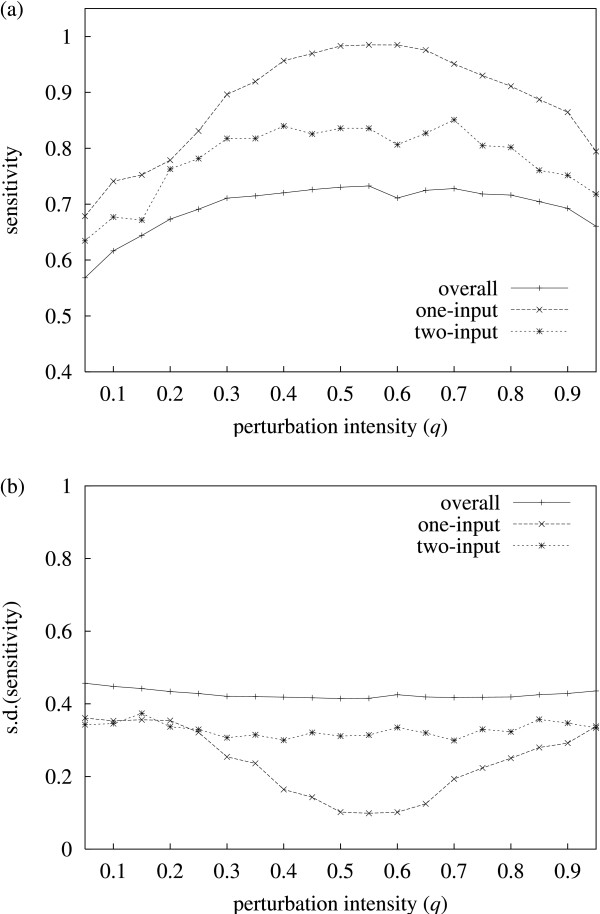
Results: It was found that a relatively small number of perturbations significantly improve inference accuracy, particularly for low-order inputs of one or two genes. The perturbations themselves should alter the expression level of approximately 50-60% of the genes in the network.
Conclusions: Time-series obtained from perturbations are a common form of expression data. This study illustrates how gene networks can be significantly reconstructed from such time-series while requiring only a relatively small number of calibrated perturbations, even for large networks, thus reducing experimental costs.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
