

The landscape of genetic complexity across 5,700 gene expression traits in yeast
- PMID: 15659551
- PMCID: PMC547855
- DOI: 10.1073/pnas.0408709102
The landscape of genetic complexity across 5,700 gene expression traits in yeast
Abstract
Many studies have identified quantitative trait loci (QTLs) that contribute to continuous variation in heritable traits of interest. However, general principles regarding the distribution of QTL numbers, effect sizes, and combined effects of multiple QTLs remain to be elucidated. Here, we characterize complex genetics underlying inheritance of thousands of transcript levels in a cross between two strains of Saccharomyces cerevisiae. Most detected QTLs have weak effects, with a median variance explained of 27% for highly heritable transcripts. Despite the high statistical power of the study, no QTLs were detected for 40% of highly heritable transcripts, indicating extensive genetic complexity. Modeling of QTL detection showed that only 3% of highly heritable transcripts are consistent with single-locus inheritance, 17-18% are consistent with control by one or two loci, and half require more than five loci under additive models. Strikingly, analysis of parent and progeny trait distributions showed that a majority of transcripts exhibit transgressive segregation. Sixteen percent of highly heritable transcripts exhibit evidence of interacting loci. Our results will aid design of future QTL mapping studies and may shed light on the evolution of quantitative traits.
Figures
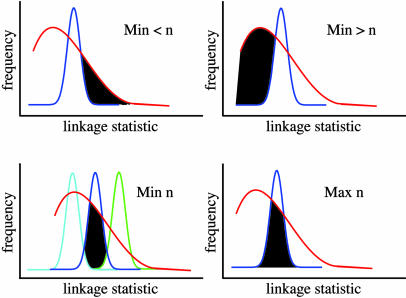
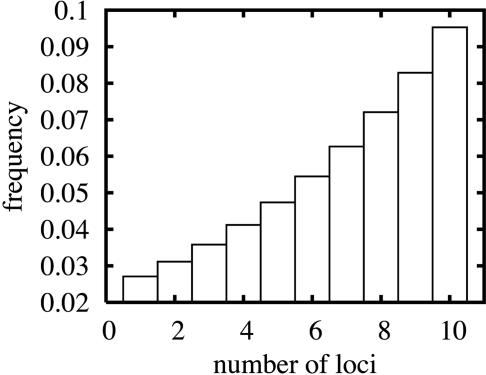
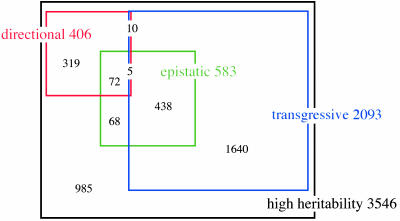
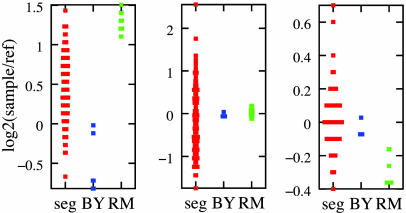
References
-
- Lynch, M. & Walsh, B. (1998) Genetics and Analysis of Quantitative Traits (Sinauer, Sunderland, MA).
-
- Flint, J. & Mott, R. (2001) Nat. Rev. Genet. 2, 437-445. - PubMed
-
- Paran, I. & Zamir, D. (2003) Trends Genet. 19, 303-306. - PubMed
-
- Glazier, A. M., Nadeau, J. H. & Aitman, T. J. (2002) Science 298, 2345-2349. - PubMed
-
- Laitinen, T., Polvi, A., Rydman, P., Vendelin, J., Pulkkinen, V., Salmikangas, P., Makela, S., Rehn, M., Pirskanen, A., Rautanen, A., et al. (2004) Science 304, 300-304. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
