

A model of the statistical power of comparative genome sequence analysis
- PMID: 15660152
- PMCID: PMC539325
- DOI: 10.1371/journal.pbio.0030010
A model of the statistical power of comparative genome sequence analysis
Abstract
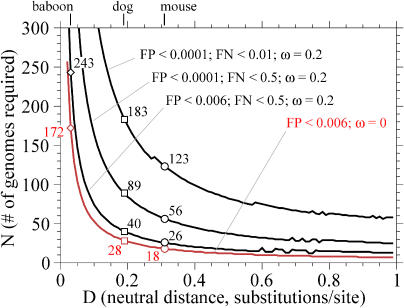
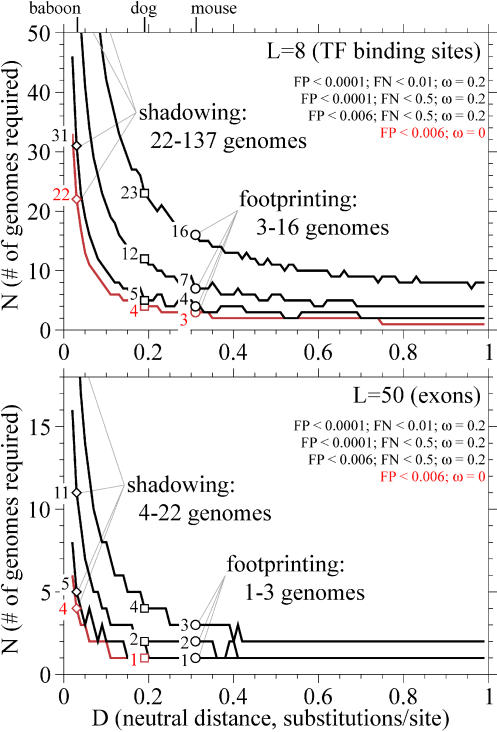
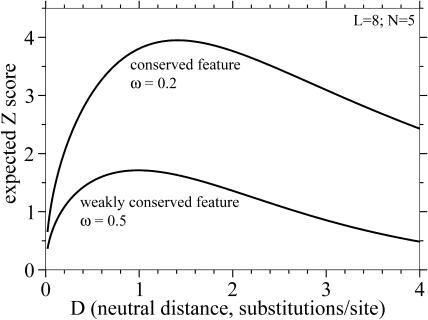
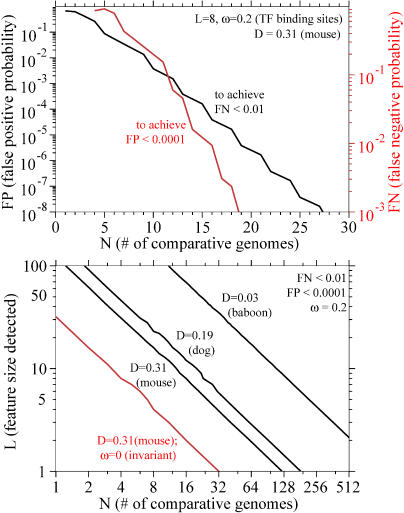
Comparative genome sequence analysis is powerful, but sequencing genomes is expensive. It is desirable to be able to predict how many genomes are needed for comparative genomics, and at what evolutionary distances. Here I describe a simple mathematical model for the common problem of identifying conserved sequences. The model leads to some useful rules of thumb. For a given evolutionary distance, the number of comparative genomes needed for a constant level of statistical stringency in identifying conserved regions scales inversely with the size of the conserved feature to be detected. At short evolutionary distances, the number of comparative genomes required also scales inversely with distance. These scaling behaviors provide some intuition for future comparative genome sequencing needs, such as the proposed use of "phylogenetic shadowing" methods using closely related comparative genomes, and the feasibility of high-resolution detection of small conserved features.
Figures
References
-
- Hardison RC. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 2000;16:369–372. - PubMed
-
- Sidow A. Sequence first. Ask questions later. Cell. 2002;111:13–16. - PubMed
-
- Thomas JW, Touchman JW, Blakesley RW, Bouffard GG, Beckstrom-Sternberg SM, et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature. 2003;424:788–793. - PubMed
-
- Cliften PF, Hillier LW, Fulton L, Graves T, Miner T, et al. Surveying Saccharomyces genomes to identify functional elements by comparative DNA sequence analysis. Genome Res. 2001;11:1175–1186. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
