

Exploring the helix-coil transition via all-atom equilibrium ensemble simulations
- PMID: 15665128
- PMCID: PMC1305346
- DOI: 10.1529/biophysj.104.051938
Exploring the helix-coil transition via all-atom equilibrium ensemble simulations
Abstract
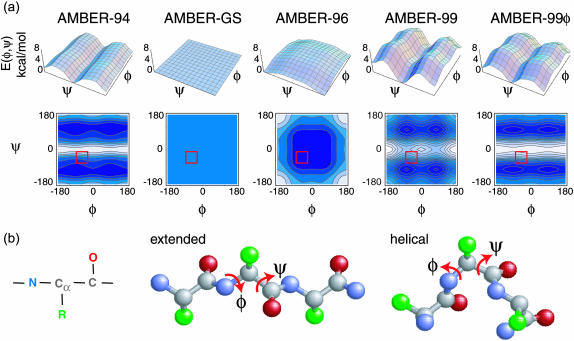
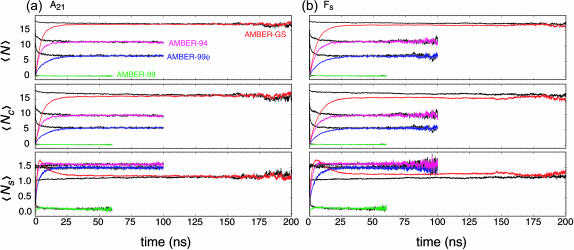
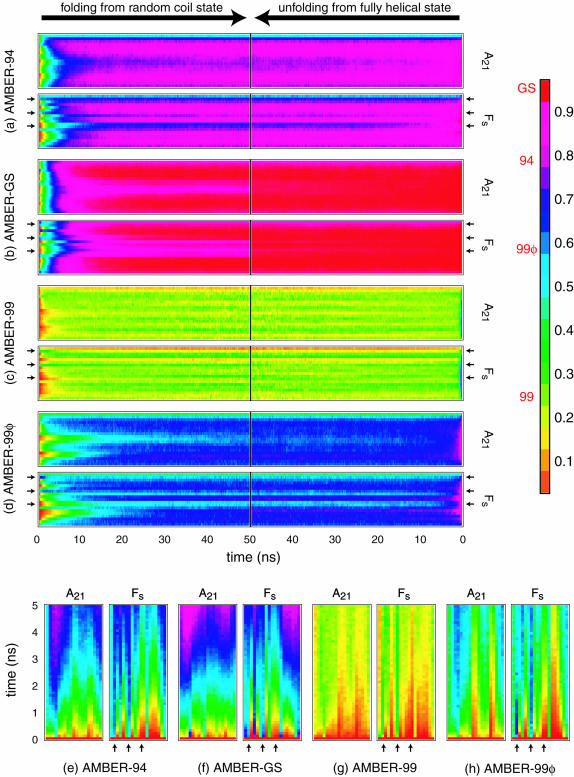
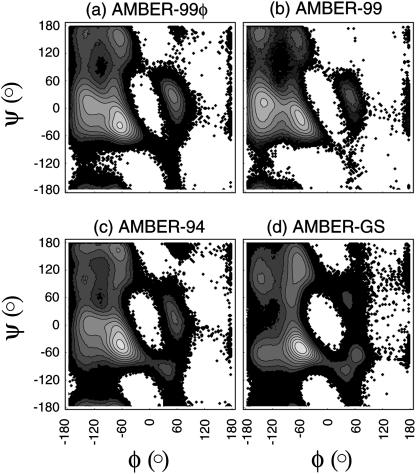
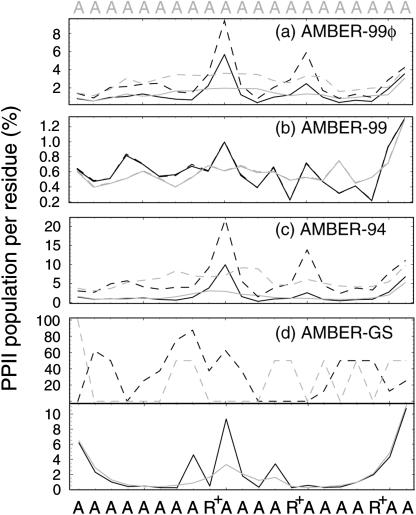
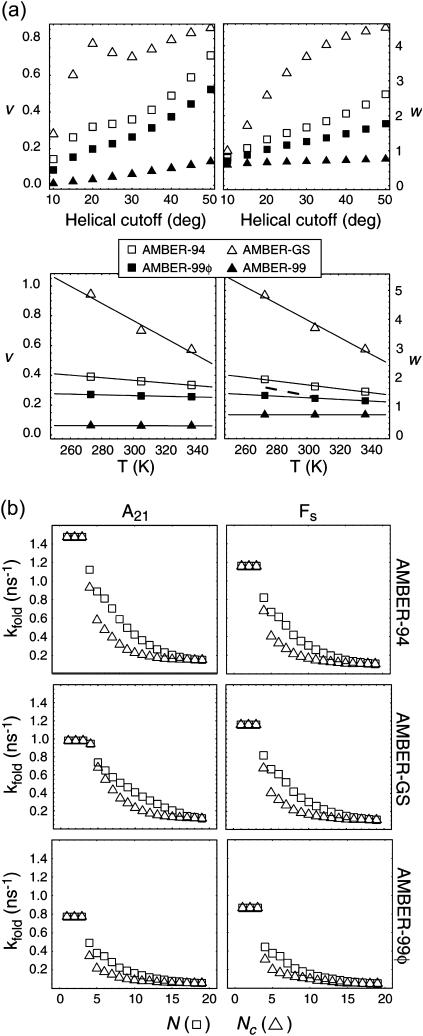
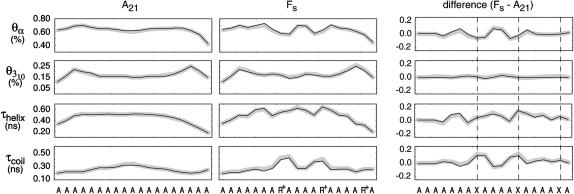
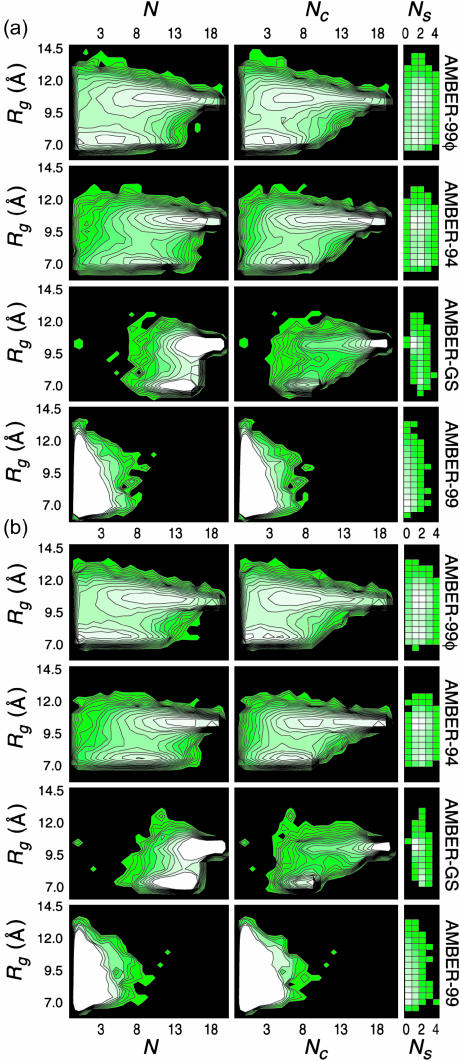
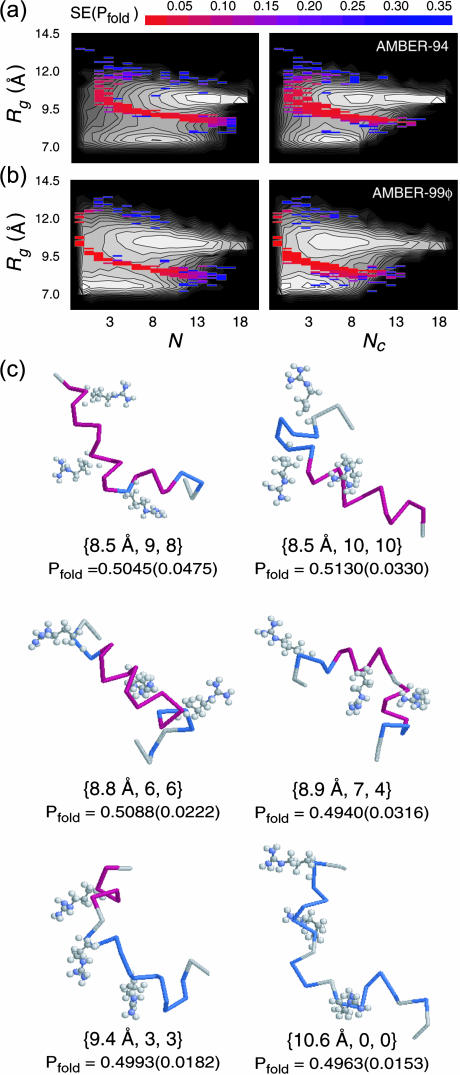
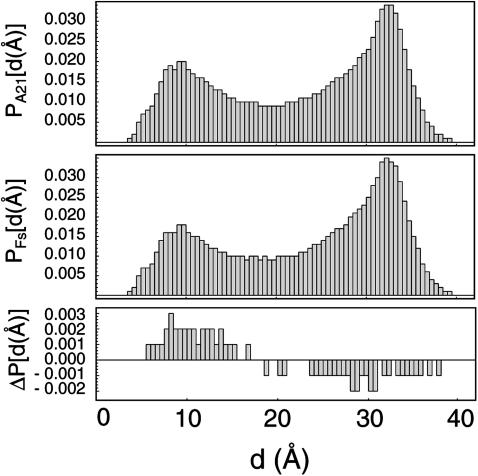
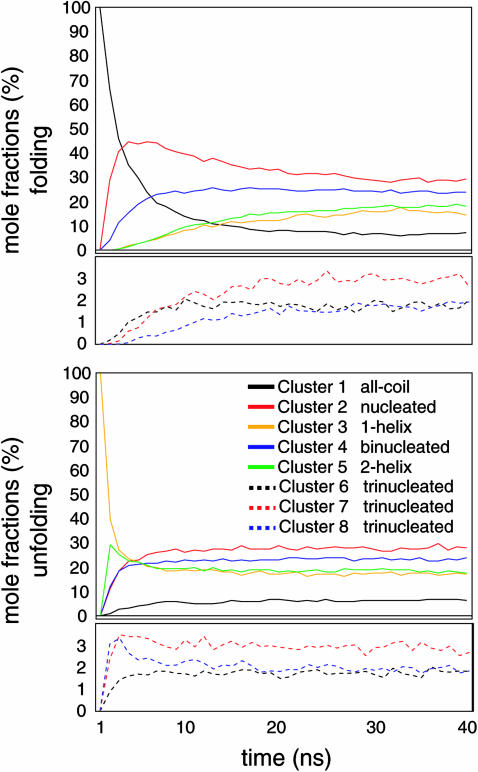
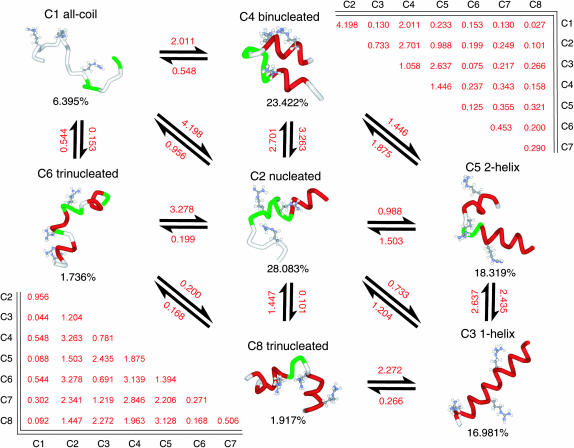
The ensemble folding of two 21-residue alpha-helical peptides has been studied using all-atom simulations under several variants of the AMBER potential in explicit solvent using a global distributed computing network. Our extensive sampling, orders of magnitude greater than the experimental folding time, results in complete convergence to ensemble equilibrium. This allows for a quantitative assessment of these potentials, including a new variant of the AMBER-99 force field, denoted AMBER-99 phi, which shows improved agreement with experimental kinetic and thermodynamic measurements. From bulk analysis of the simulated AMBER-99 phi equilibrium, we find that the folding landscape is pseudo-two-state, with complexity arising from the broad, shallow character of the "native" and "unfolded" regions of the phase space. Each of these macrostates allows for configurational diffusion among a diverse ensemble of conformational microstates with greatly varying helical content and molecular size. Indeed, the observed structural dynamics are better represented as a conformational diffusion than as a simple exponential process, and equilibrium transition rates spanning several orders of magnitude are reported. After multiple nucleation steps, on average, helix formation proceeds via a kinetic "alignment" phase in which two or more short, low-entropy helical segments form a more ideal, single-helix structure.
Figures
Similar articles
-
Helix nucleation kinetics from molecular simulations in explicit solvent.Proteins. 2001 Jan 1;42(1):77-84. Proteins. 2001. PMID: 11093262
-
Peptide folding kinetics from replica exchange molecular dynamics.Phys Rev E Stat Nonlin Soft Matter Phys. 2008 Mar;77(3 Pt 1):030902. doi: 10.1103/PhysRevE.77.030902. Epub 2008 Mar 24. Phys Rev E Stat Nonlin Soft Matter Phys. 2008. PMID: 18517321
-
Kinetics of the coil-to-helix transition on a rough energy landscape.Phys Rev E Stat Nonlin Soft Matter Phys. 2003 Nov;68(5 Pt 1):051901. doi: 10.1103/PhysRevE.68.051901. Epub 2003 Nov 3. Phys Rev E Stat Nonlin Soft Matter Phys. 2003. PMID: 14682814
-
Modeling loop entropy.Methods Enzymol. 2011;487:99-132. doi: 10.1016/B978-0-12-381270-4.00004-4. Methods Enzymol. 2011. PMID: 21187223 Free PMC article. Review.
-
Enhanced conformational sampling to visualize a free-energy landscape of protein complex formation.Biochem J. 2016 Jun 15;473(12):1651-62. doi: 10.1042/BCJ20160053. Biochem J. 2016. PMID: 27288028 Free PMC article. Review.
Cited by
-
Elastic network normal modes provide a basis for protein structure refinement.J Chem Phys. 2012 May 21;136(19):195101. doi: 10.1063/1.4710986. J Chem Phys. 2012. PMID: 22612113 Free PMC article.
-
Molecular dynamics simulations on aqueous two-phase systems - Single PEG-molecules in solution.BMC Biophys. 2012 Aug 8;5:14. doi: 10.1186/2046-1682-5-14. BMC Biophys. 2012. PMID: 22873343 Free PMC article.
-
Motional timescale predictions by molecular dynamics simulations: case study using proline and hydroxyproline sidechain dynamics.Proteins. 2014 Feb;82(2):195-215. doi: 10.1002/prot.24350. Epub 2013 Sep 17. Proteins. 2014. PMID: 23818175 Free PMC article.
-
Predictions of the Poses and Affinity of a Ligand over the Entire Surface of a NEET Protein: The Case of Human MitoNEET.J Chem Inf Model. 2023 Jan 23;63(2):643-654. doi: 10.1021/acs.jcim.2c01280. Epub 2023 Jan 9. J Chem Inf Model. 2023. PMID: 36623826 Free PMC article.
-
Comparative biochemical and computational study of the role of naturally occurring mutations at Ambler positions 104 and 170 in GES β-lactamases.Antimicrob Agents Chemother. 2010 Nov;54(11):4864-71. doi: 10.1128/AAC.00771-10. Epub 2010 Aug 9. Antimicrob Agents Chemother. 2010. PMID: 20696873 Free PMC article.
References
-
- Banavar, J. R., A. Maritan, C. Micheletti, and A. Trovato. 2002. Geometry and physics of proteins. Proteins. 47:315–322. - PubMed
-
- Berendsen, H., J. Postma, W. Vangunsteren, A. Dinola, and J. Haak. 1984. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690.
-
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimisation, and dynamics calculations. J. Comput. Chem. 4:187–217.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
