

Bayesian and maximum likelihood phylogenetic analyses of protein sequence data under relative branch-length differences and model violation
- PMID: 15676079
- PMCID: PMC549035
- DOI: 10.1186/1471-2148-5-8
Bayesian and maximum likelihood phylogenetic analyses of protein sequence data under relative branch-length differences and model violation
Abstract
Background: Bayesian phylogenetic inference holds promise as an alternative to maximum likelihood, particularly for large molecular-sequence data sets. We have investigated the performance of Bayesian inference with empirical and simulated protein-sequence data under conditions of relative branch-length differences and model violation.
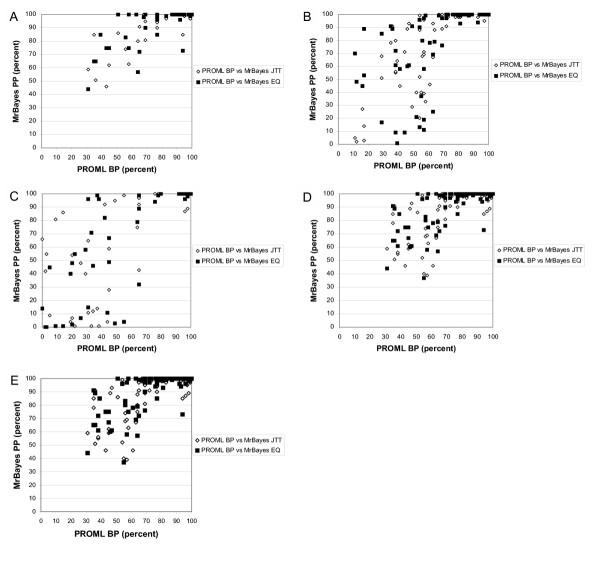
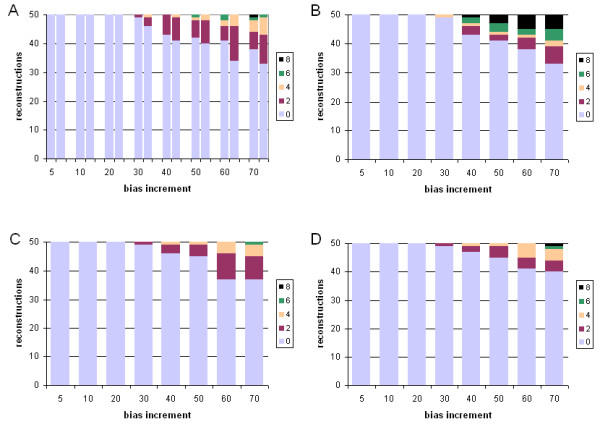
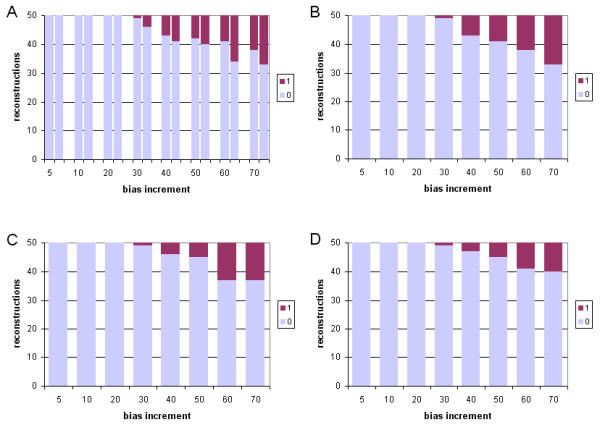
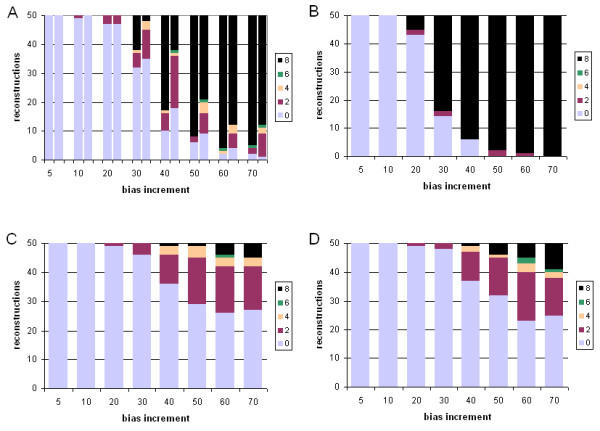
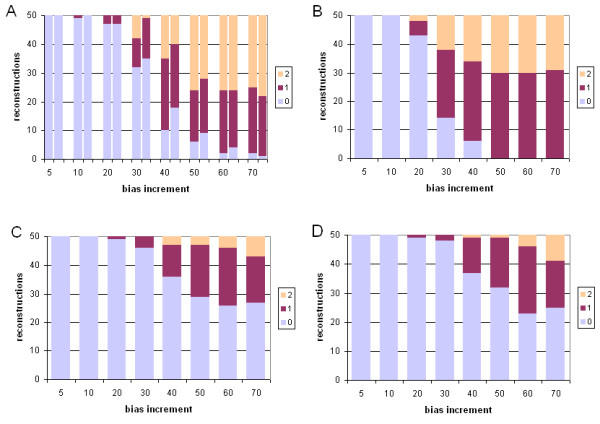
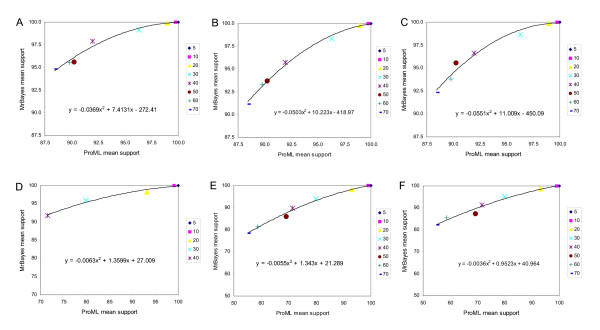
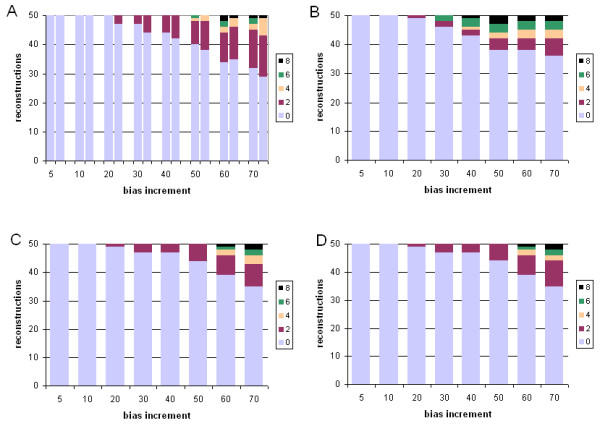
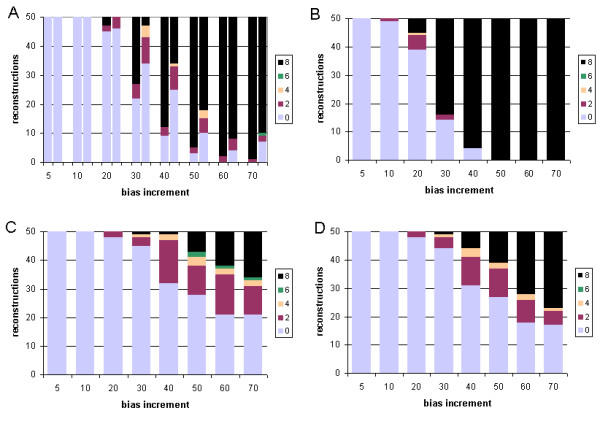
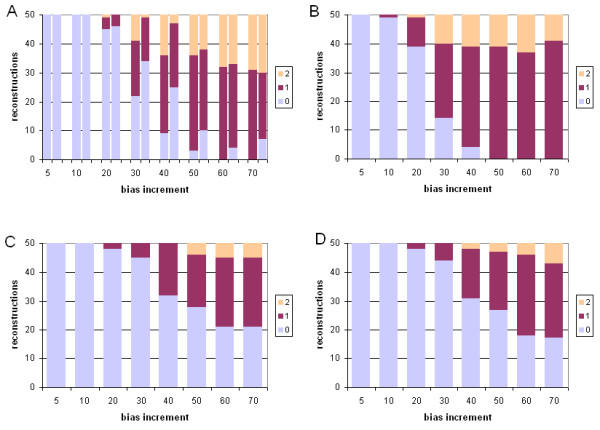
Results: With empirical protein-sequence data, Bayesian posterior probabilities provide more-generous estimates of subtree reliability than does the nonparametric bootstrap combined with maximum likelihood inference, reaching 100% posterior probability at bootstrap proportions around 80%. With simulated 7-taxon protein-sequence datasets, Bayesian posterior probabilities are somewhat more generous than bootstrap proportions, but do not saturate. Compared with likelihood, Bayesian phylogenetic inference can be as or more robust to relative branch-length differences for datasets of this size, particularly when among-sites rate variation is modeled using a gamma distribution. When the (known) correct model was used to infer trees, Bayesian inference recovered the (known) correct tree in 100% of instances in which one or two branches were up to 20-fold longer than the others. At ratios more extreme than 20-fold, topological accuracy of reconstruction degraded only slowly when only one branch was of relatively greater length, but more rapidly when there were two such branches. Under an incorrect model of sequence change, inaccurate trees were sometimes observed at less extreme branch-length ratios, and (particularly for trees with single long branches) such trees tended to be more inaccurate. The effect of model violation on accuracy of reconstruction for trees with two long branches was more variable, but gamma-corrected Bayesian inference nonetheless yielded more-accurate trees than did either maximum likelihood or uncorrected Bayesian inference across the range of conditions we examined. Assuming an exponential Bayesian prior on branch lengths did not improve, and under certain extreme conditions significantly diminished, performance. The two topology-comparison metrics we employed, edit distance and Robinson-Foulds symmetric distance, yielded different but highly complementary measures of performance.
Conclusions: Our results demonstrate that Bayesian inference can be relatively robust against biologically reasonable levels of relative branch-length differences and model violation, and thus may provide a promising alternative to maximum likelihood for inference of phylogenetic trees from protein-sequence data.
Figures

References
-
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. - PubMed
-
- Felsenstein J. Inferring phylogenies. Sunderland MA: Sinauer Associates; 2004.
-
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. - PubMed
-
- Kishino H, Miyata T, Hasegawa M. Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J Mol Evol. 1990;31:151–160.
-
- Hasegawa M, Kishino H. Accuracies of the simple methods for estimating the bootstrap probability of a maximum-likelihood tree. Mol Biol Evol. 1994;11:142–145.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
