

Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains
- PMID: 15677316
- PMCID: PMC548970
- DOI: 10.1073/pnas.0408885102
Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains
Abstract
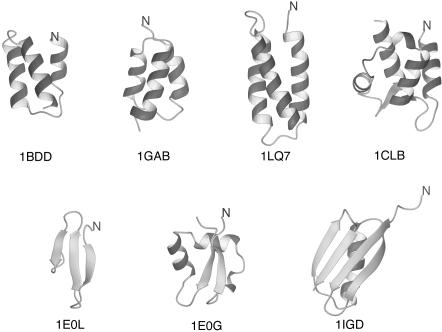
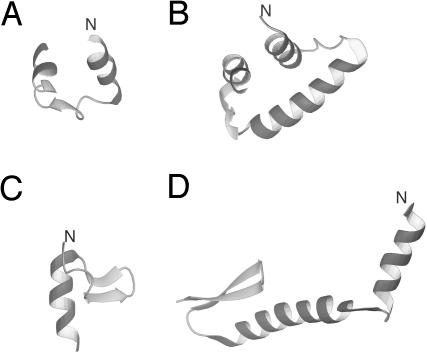
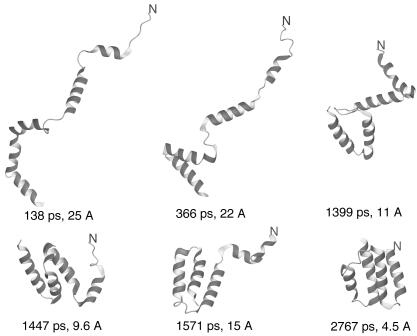
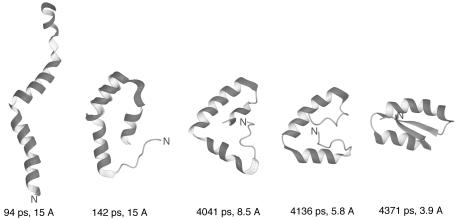
We report the application of Langevin dynamics to the physics-based united-residue (UNRES) force field developed in our laboratory. Ten trajectories were run on seven proteins [PDB ID codes 1BDD (alpha; 46 residues), 1GAB (alpha; 47 residues), 1LQ7 (alpha; 67 residues), 1CLB (alpha; 75 residues), 1E0L (beta; 28 residues), and 1E0G (alpha+beta; 48 residues), and 1IGD (alpha+beta; 61 residues)] with the UNRES force field parameterized by using our recently developed method for obtaining a hierarchical structure of the energy landscape. All alpha-helical proteins and 1E0G folded to the native-like structures, whereas 1IGD and 1E0L yielded mostly nonnative alpha-helical folds although the native-like structures are lowest in energy for these two proteins, which can be attributed to neglecting the entropy factor in the current parameterization of UNRES. Average folding times for successful folding simulations were of the order of nanoseconds, whereas even the ultrafast-folding proteins fold only in microseconds, which implies that the UNRES time scale is approximately three orders of magnitude larger than the experimental time scale because the fast motions of the secondary degrees of freedom are averaged out. Folding with Langevin dynamics required 2-10 h of CPU time on average with a single AMD Athlon MP 2800+ processor depending on the size of the protein. With the advantage of parallel processing, this process leads to the possibility to explore thousands of folding pathways and to predict not only the native structure but also the folding scenario of a protein together with its quantitative kinetic and thermodynamic characteristics.
Figures
Comment in
-
Putting the pathway back into protein folding.Proc Natl Acad Sci U S A. 2005 Feb 15;102(7):2265-6. doi: 10.1073/pnas.0500128102. Epub 2005 Feb 9. Proc Natl Acad Sci U S A. 2005. PMID: 15703287 Free PMC article. No abstract available.
References
-
- Scheraga, H. A., Liwo, A., Ołdziej, S., Czaplewski, C., Pillardy, J., Ripoll, D. R., Vila, J. A., Kazmierkiewicz, R., Saunders, J. A., Arnautova, Y. A., et al. (2004) Front. Biosci. 9, 3296-3323. - PubMed
-
- Skolnick, J., Zhang, Y., Arakaki, A. K., Kolinski, A., Boniecki, M., Szilagyi, A. & Kihara, D. (2003) Proteins Struct. Funct. Genet. 53, Suppl. 6, 469-479. - PubMed
-
- Bradley, P., Chivian, D., Meiler, J., Misura, K. M. S., Rohl, C. A., Schief, W. R., Wedemeyer, W. J., Schueler-Furman, O., Murphy, P., Schonbrun, J., et al. (2003) Proteins Struct. Funct. Genet. 53, Suppl. 6, 457-468. - PubMed
-
- Day, R. & Daggett, V. (2003) Adv. Protein Chem. 66, 373-403. - PubMed
-
- Fersht, A. R. & Daggett, V. (2002) Cell 108, 573-582. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
