

Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1)
- PMID: 15681471
- PMCID: PMC1867514
- DOI: 10.1016/S1525-1578(10)60005-0
Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1)
Abstract
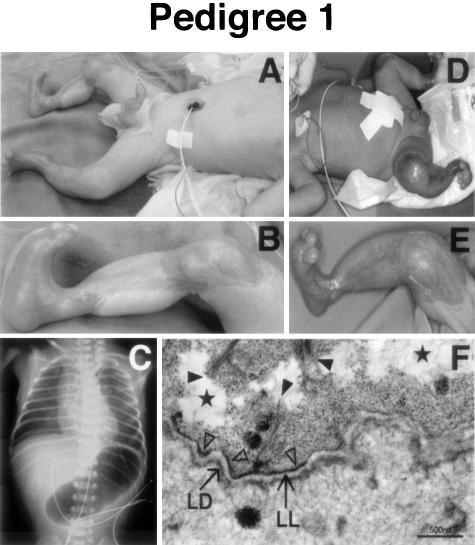
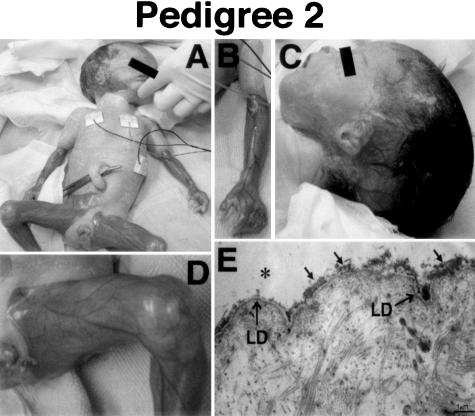
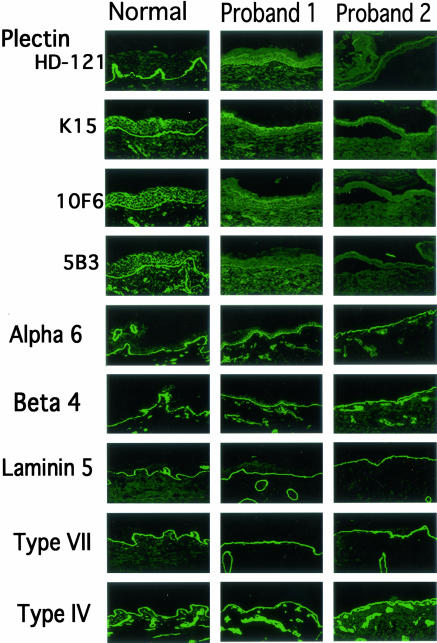
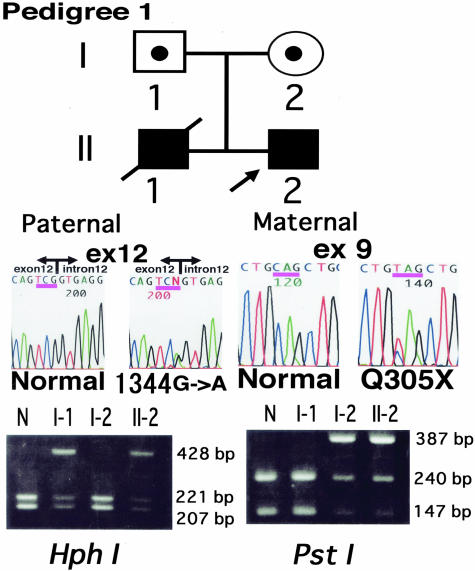
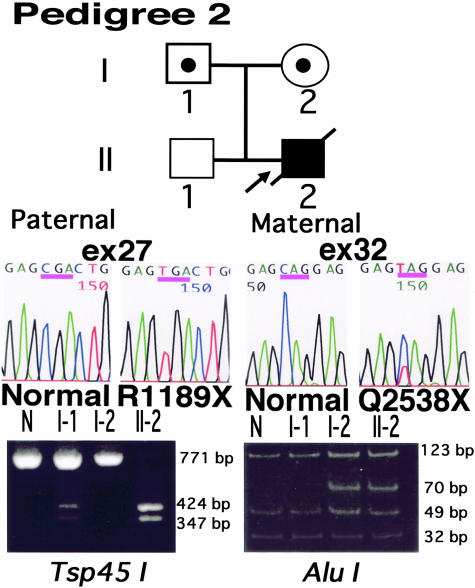
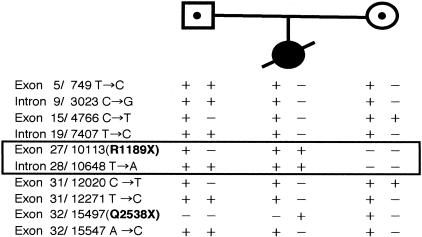
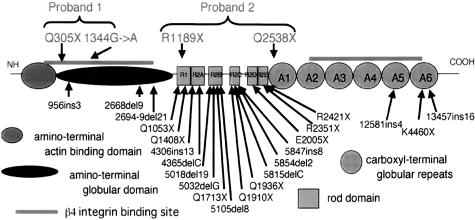
Epidermolysis bullosa (EB) is an inherited mechano-bullous disorder of the skin, and is divided into three major categories: EB simplex (EBS), dystrophic EB, and junctional EB (JEB). Mutations in the plectin gene (PLEC1) cause EBS associated with muscular dystrophy, whereas JEB associated with pyloric atresia (PA) results from mutations in the alpha6 and beta4 integrin genes. In this study, we examined three EB patients associated with PA from two distinct families. Electron microscopy detected blister formation within the basal keratinocytes leading to the diagnosis of EBS. Surprisingly, immunohistochemical studies using monoclonal antibodies to a range of basement membrane proteins showed that the expression of plectin was absent or markedly attenuated. Sequence analysis demonstrated four novel PLEC1 mutations. One proband was a compound heterozygote for a nonsense mutation of Q305X and a splice-site mutation of 1344G-->A. An exon-trapping experiment suggested that the splice-site mutation induced aberrant splicing of the gene. The second proband harbored a heterozygous maternal nonsense mutation, Q2538X and homozygous nonsense mutations R1189X. Analysis of the intragenic polymorphisms of PLEC1 suggested that R1189X mutations were due to paternal segmental uniparental isodisomy. These results indicate that PLEC1 is a possible causative gene in this clinical subtype, EBS associated with PA. Furthermore, two patients out of our three cases died in infancy. In terms of clinical prognosis, this novel subtype is the lethal variant in the EBS category.
Figures
Similar articles
-
Plectin expression patterns determine two distinct subtypes of epidermolysis bullosa simplex.Hum Mutat. 2010 Mar;31(3):308-16. doi: 10.1002/humu.21189. Hum Mutat. 2010. PMID: 20052759
-
DNA-based prenatal diagnosis of plectin-deficient epidermolysis bullosa simplex associated with pyloric atresia.Int J Dermatol. 2011 Apr;50(4):439-42. doi: 10.1111/j.1365-4632.2010.04771.x. Int J Dermatol. 2011. PMID: 21413955
-
A homozygous nonsense mutation in the PLEC1 gene in patients with epidermolysis bullosa simplex with muscular dystrophy.J Clin Invest. 1996 Nov 15;98(10):2196-200. doi: 10.1172/JCI119028. J Clin Invest. 1996. PMID: 8941634 Free PMC article.
-
Progress in epidermolysis bullosa: the phenotypic spectrum of plectin mutations.Exp Dermatol. 2005 Apr;14(4):241-9. doi: 10.1111/j.0906-6705.2005.00324.x. Exp Dermatol. 2005. PMID: 15810881 Review.
-
Differential expression of pyloric atresia in junctional epidermolysis bullosa with ITGB4 mutations suggests that pyloric atresia is due to factors other than the mutations and not predictive of a poor outcome: three novel mutations and a review of the literature.Acta Derm Venereol. 2008;88(5):438-48. doi: 10.2340/00015555-0484. Acta Derm Venereol. 2008. PMID: 18779879 Review.
Cited by
-
Diseases of epidermal keratins and their linker proteins.Exp Cell Res. 2007 Jun 10;313(10):1995-2009. doi: 10.1016/j.yexcr.2007.03.029. Epub 2007 Apr 24. Exp Cell Res. 2007. PMID: 17531221 Free PMC article. Review.
-
Compound Heterozygous Mutations with a Novel Variant in Integrin Beta4 Cause Epidermolysis Bullosa with Pyloric Atresia and Urologic Abnormalities.Yonsei Med J. 2020 Sep;61(9):831-833. doi: 10.3349/ymj.2020.61.9.831. Yonsei Med J. 2020. PMID: 32882768 Free PMC article. No abstract available.
-
Epidermolysis Bullosa With Congenital Absence of Skin: Congenital Corneal Cloudiness and Esophagogastric Obstruction Including Extended Genotypic Spectrum of PLEC, LAMC2, ITGB4 and COL7A1.Front Genet. 2022 Apr 1;13:847150. doi: 10.3389/fgene.2022.847150. eCollection 2022. Front Genet. 2022. PMID: 35432467 Free PMC article.
-
Case report: A case of epidermolysis bullosa complicated with pyloric atresia and a literature review.Front Pediatr. 2023 Mar 23;11:1098273. doi: 10.3389/fped.2023.1098273. eCollection 2023. Front Pediatr. 2023. PMID: 37033187 Free PMC article.
-
Targeted proteolysis of plectin isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna.PLoS Genet. 2011 Dec;7(12):e1002396. doi: 10.1371/journal.pgen.1002396. Epub 2011 Dec 1. PLoS Genet. 2011. PMID: 22144912 Free PMC article.
References
-
- Fine JD, Eady RA, Bauer EA, Briggaman RA, Bruckner-Tuderman L, Christiano A, Heagerty A, Hintner H, Jonkman MF, McGrath J, McGuire J, Moshell A, Shimizu H, Tadini G, Uitto J. Revised classification system for inherited epidermolysis bullosa: report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2000;42:1051–66. - PubMed
-
- Pulkkinen L, Uitto J. Mutations analysis and molecular genetics of epidermolysis bullosa. Matrix Biol. 1999;18:29–42. - PubMed
-
- Vidal F, Aberdam D, Miquel C, Christiano AM, Pulkkinen L, Uitto J, Ortonne JP, Meneguzzi G. Integrin beta 4 mutations associated with junctional epidermolysis bullosa with pyloric atresia. Nat Genet. 1995;10:229–234. - PubMed
-
- Pulkkinen L, Kimonis VE, Xu Y, Spanou EN, McLean WH, Uitto J. Homozygous alpha6 integrin mutation in junctional epidermolysis bullosa with congenital duodenal atresia. Hum Mol Genet. 1997;6:669–674. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
