

Role of adenosine A1 and A3 receptors in regulation of cardiomyocyte homeostasis after mitochondrial respiratory chain injury
- PMID: 15681707
- PMCID: PMC3457058
- DOI: 10.1152/ajpheart.01157.2004
Role of adenosine A1 and A3 receptors in regulation of cardiomyocyte homeostasis after mitochondrial respiratory chain injury
Abstract
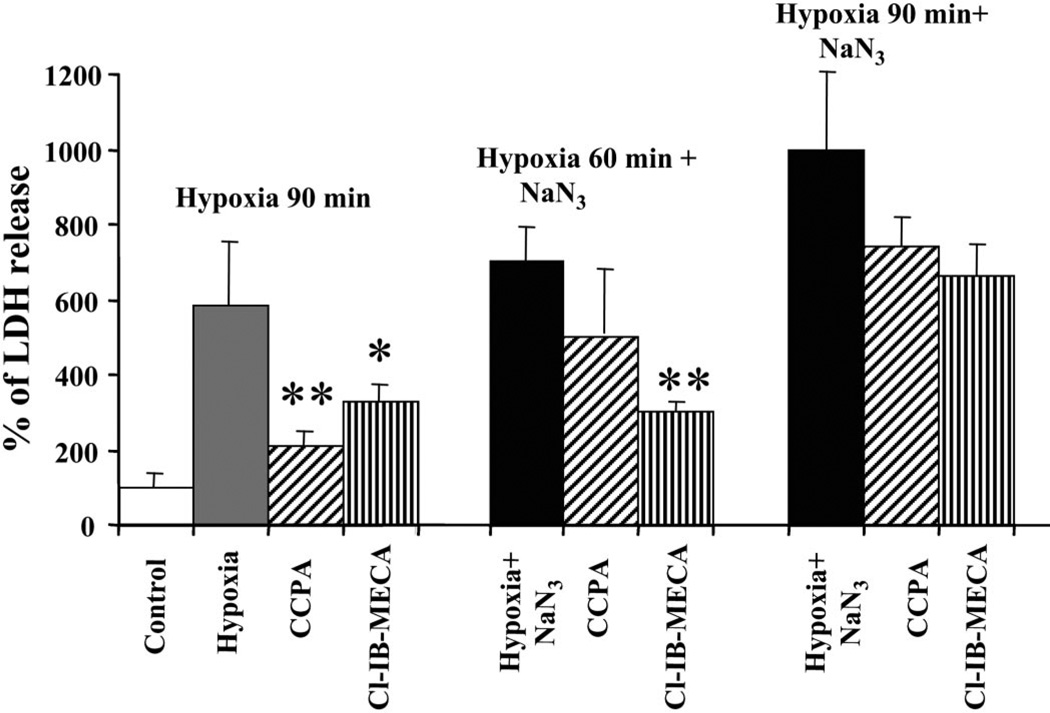
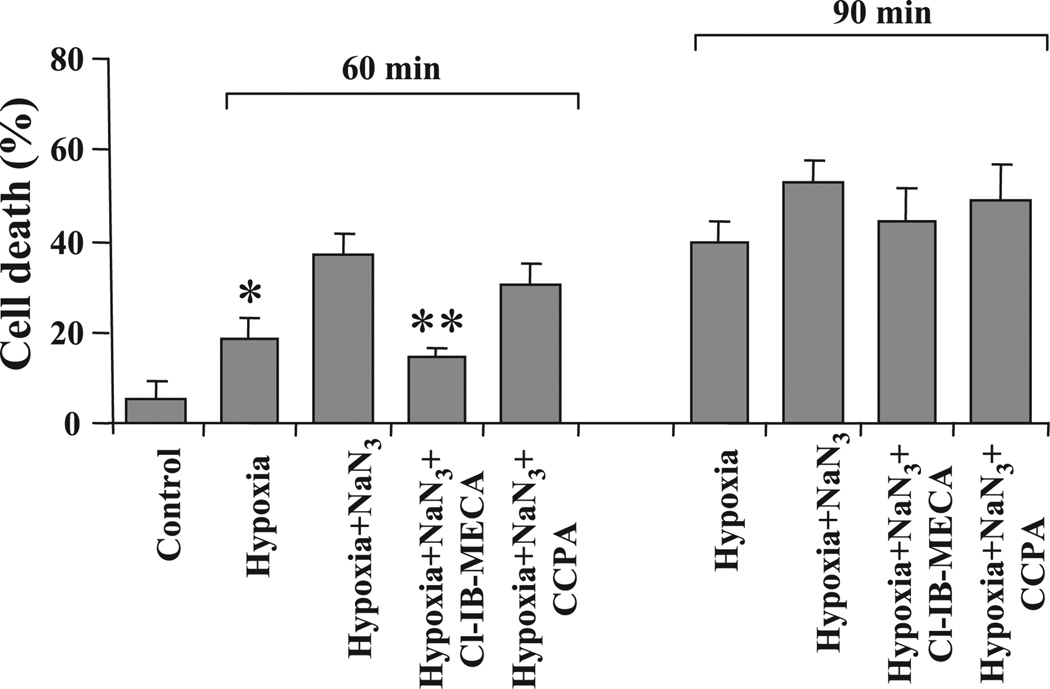
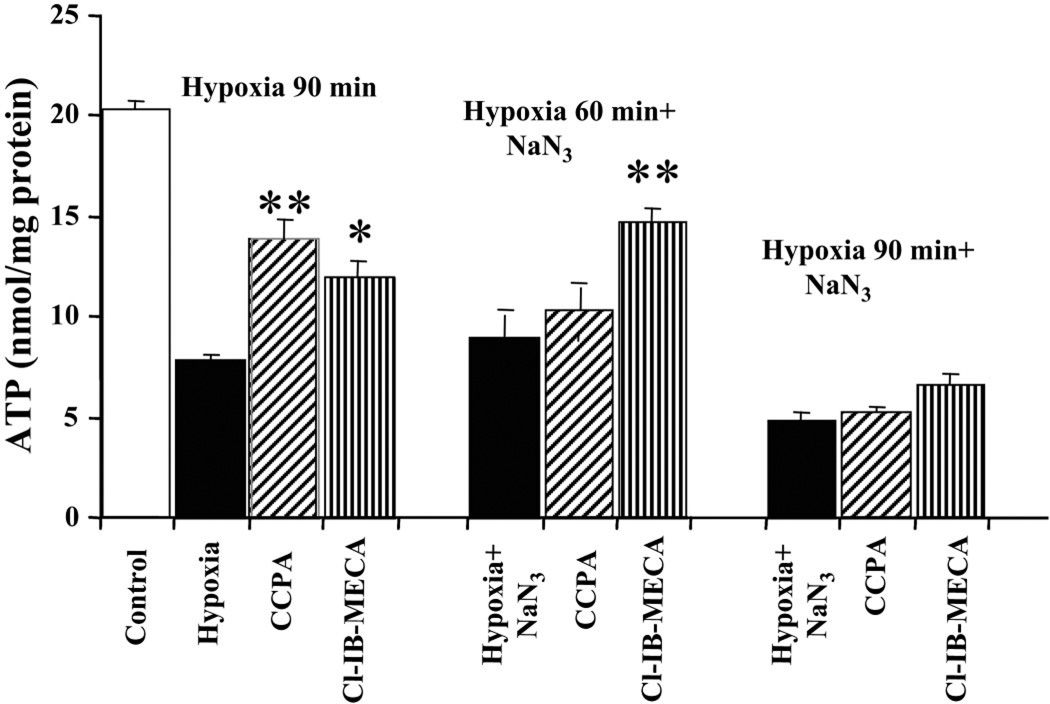
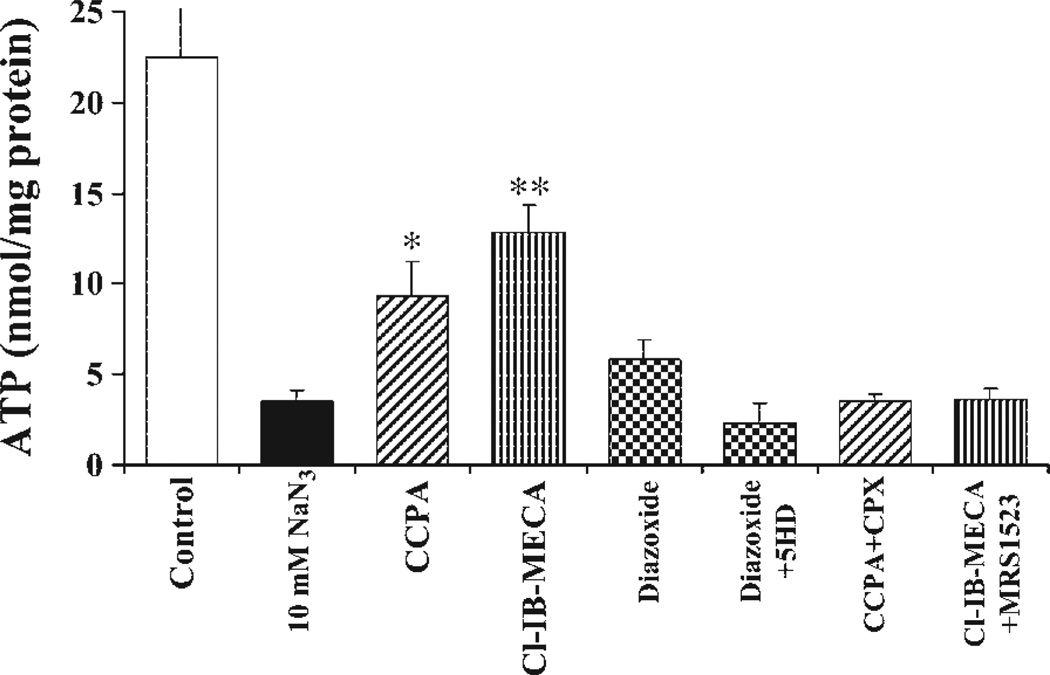
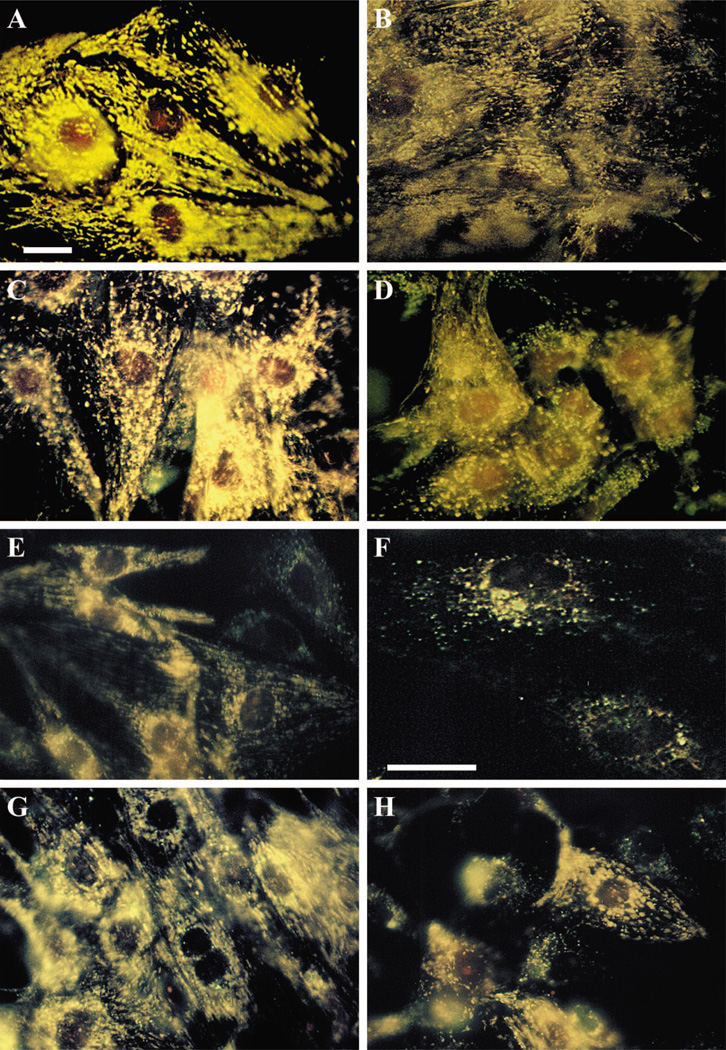
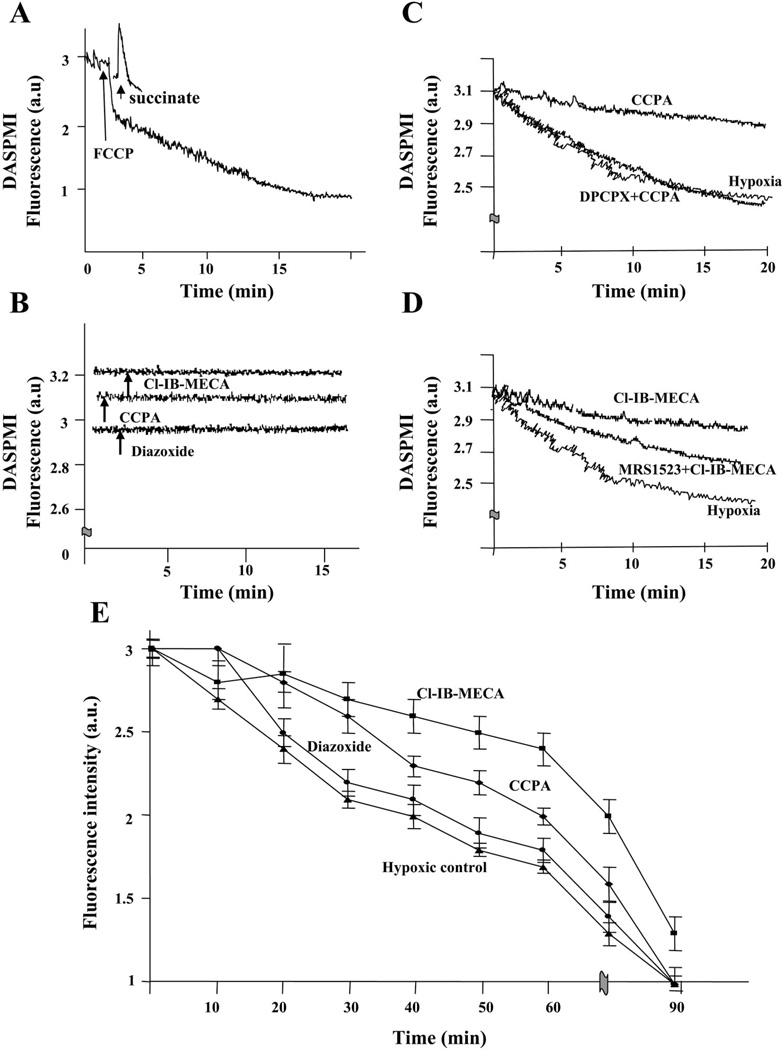
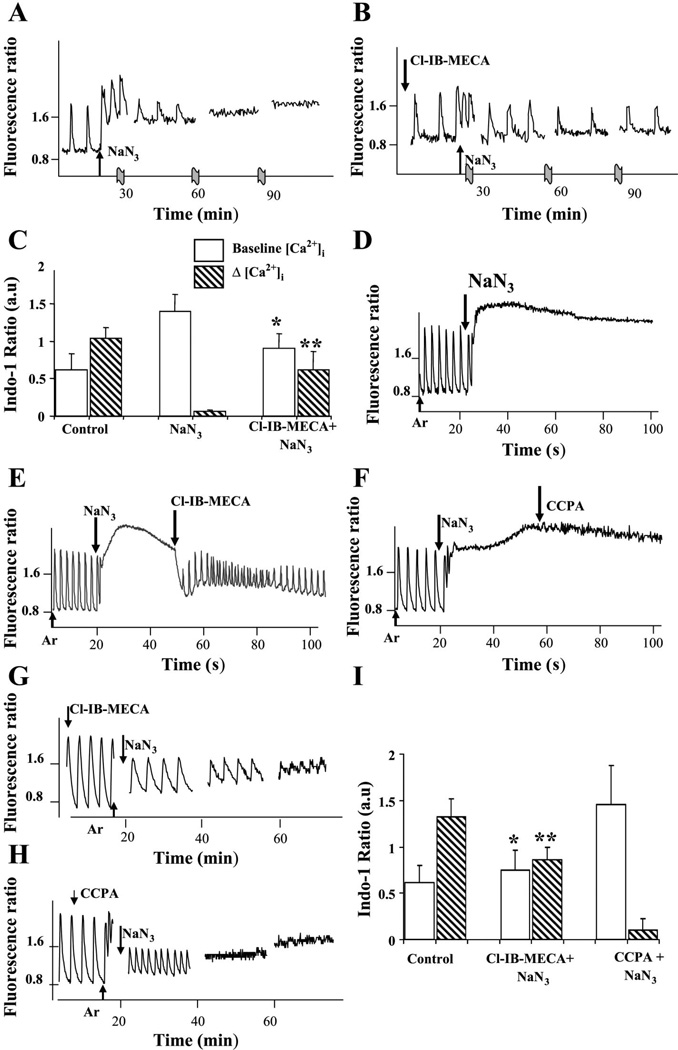
Activation of either the A(1) or the A(3) adenosine receptor (A(1)R or A(3)R, respectively) elicits delayed cardioprotection against infarction, ischemia, and hypoxia. Mitochondrial contribution to the progression of cardiomyocyte injury is well known; however, the protective effects of adenosine receptor activation in cardiac cells with a respiratory chain deficiency are poorly elucidated. The aim of our study was to further define the role of A(1)R and A(3)R activation on functional tolerance after inhibition of the terminal link of the mitochondrial respiratory chain with sodium azide, in a state of normoxia or hypoxia, compared with the effects of the mitochondrial ATP-sensitive K(+) channel opener diazoxide. Treatment with 10 mM sodium azide for 2 h in normoxia caused a considerable decrease in the total ATP level; however, activation of adenosine receptors significantly attenuated this decrease. Diazoxide (100 muM) was less effective in protection. During treatment of cultured cardiomyocytes with hypoxia in the presence of 1 mM sodium azide, the A(1)R agonist 2-chloro-N(6)-cyclopentyladenosine was ineffective, whereas the A(3)R agonist 2-chloro-N(6)-iodobenzyl-5'-N-methylcarboxamidoadenosine (Cl-IB-MECA) attenuated the decrease in ATP level and prevented cell injury. Cl-IB-MECA delayed the dissipation in the mitochondrial membrane potential during hypoxia in cells impaired in the mitochondrial respiratory chain. In cells with elevated intracellular Ca(2+) concentration after hypoxia and treatment with NaN(3) or after application of high doses of NaN(3), Cl-IB-MECA immediately decreased the elevated intracellular Ca(2+) concentration toward the diastolic control level. The A(1)R agonist was ineffective. This may be especially important for the development of effective pharmacological agents, because mitochondrial dysfunction is a leading factor in the pathophysiological cascade of heart disease.
Figures
Similar articles
-
Adenosine A(1) and A (3) receptor agonists reduce hypoxic injury through the involvement of P38 MAPK.Mol Cell Biochem. 2010 Dec;345(1-2):153-60. doi: 10.1007/s11010-010-0568-5. Epub 2010 Aug 22. Mol Cell Biochem. 2010. PMID: 20730620
-
Induction of apoptosis in rat cardiocytes by A3 adenosine receptor activation and its suppression by isoproterenol.Exp Cell Res. 2000 May 25;257(1):111-26. doi: 10.1006/excr.2000.4882. Exp Cell Res. 2000. PMID: 10854059 Free PMC article.
-
Role of adenosine receptor activation in antioxidant enzyme regulation during ischemia-reperfusion in the isolated rat heart.Antioxid Redox Signal. 2004 Apr;6(2):335-44. doi: 10.1089/152308604322899404. Antioxid Redox Signal. 2004. PMID: 15025935
-
Activation of A(3)adenosine receptor protects against doxorubicin-induced cardiotoxicity.J Mol Cell Cardiol. 2001 Jun;33(6):1249-61. doi: 10.1006/jmcc.2001.1387. J Mol Cell Cardiol. 2001. PMID: 11444927 Free PMC article.
-
The mitochondrial K(ATP) channel and cardioprotection.Ann Thorac Surg. 2003 Feb;75(2):S667-73. doi: 10.1016/s0003-4975(02)04689-1. Ann Thorac Surg. 2003. PMID: 12607710 Review.
Cited by
-
Adenosine receptors as therapeutic targets.Nat Rev Drug Discov. 2006 Mar;5(3):247-64. doi: 10.1038/nrd1983. Nat Rev Drug Discov. 2006. PMID: 16518376 Free PMC article. Review.
-
Adenosine A(1) and A (3) receptor agonists reduce hypoxic injury through the involvement of P38 MAPK.Mol Cell Biochem. 2010 Dec;345(1-2):153-60. doi: 10.1007/s11010-010-0568-5. Epub 2010 Aug 22. Mol Cell Biochem. 2010. PMID: 20730620
-
G protein coupled receptors signaling pathways implicate in inflammatory and immune response of rheumatoid arthritis.Inflamm Res. 2017 May;66(5):379-387. doi: 10.1007/s00011-016-1011-5. Epub 2016 Nov 23. Inflamm Res. 2017. PMID: 27878579 Review.
-
Endless Journey of Adenosine Signaling in Cardioprotective Mechanism of Conditioning Techniques: Clinical Evidence.Curr Cardiol Rev. 2023;19(6):56-71. doi: 10.2174/1573403X19666230612112259. Curr Cardiol Rev. 2023. PMID: 37309766 Free PMC article. Review.
-
Conditioned medium from hypoxic cells protects cardiomyocytes against ischemia.Mol Cell Biochem. 2012 Apr;363(1-2):167-78. doi: 10.1007/s11010-011-1169-7. Epub 2011 Dec 8. Mol Cell Biochem. 2012. PMID: 22160856
References
-
- Auchampach JA, Bolli R. Adenosine receptor subtypes in the heart: therapeutic opportunities and challenges. Am J Physiol Heart Circ Physiol. 1999;276:H1113–H1116. - PubMed
-
- Chen SJ, Bradley ME, Lee TC. Chemical hypoxia triggers apoptosis of cultured neonatal rat cardiac myocytes: modulation by calcium-regulated proteases and protein kinases. Mol Cell Biochem. 1998;178:141–149. - PubMed
-
- Downey JM. ISHR Satellite: Cellular Injury in Ischaemia. Kruger National Park, South Africa: Berg en Dal; 2004. Aug 13–16, The cellular mechanisms of ischemic and pharmacological preconditioning (Abstract 13)
-
- Dzeja PP, Bast P, Ozcan C, Valverde A, Holmuhamedov EL, Van Wylen DG, Terzic A. Targeting nucleotide-requiring enzymes: implications for diazoxide-induced cardioprotection. Am J Physiol Heart Circ Physiol. 2003;284:H1048–H1056. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous