

Neutral microepidemic evolution of bacterial pathogens
- PMID: 15684071
- PMCID: PMC548543
- DOI: 10.1073/pnas.0406993102
Neutral microepidemic evolution of bacterial pathogens
Abstract
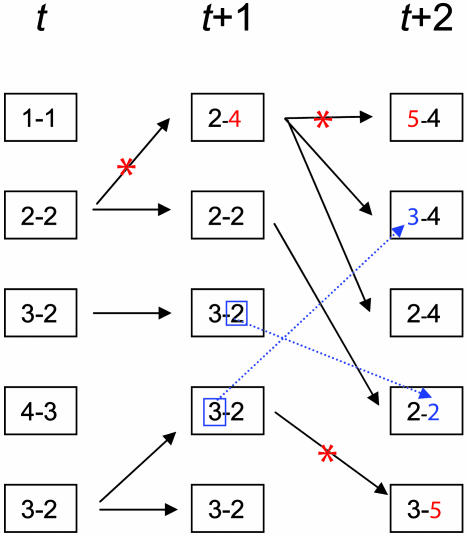
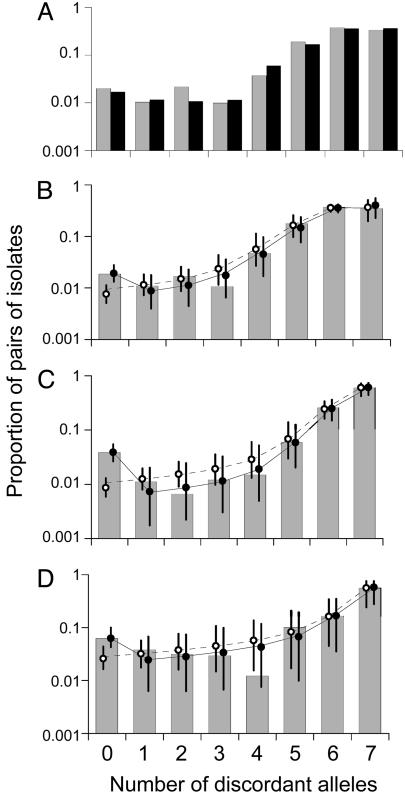
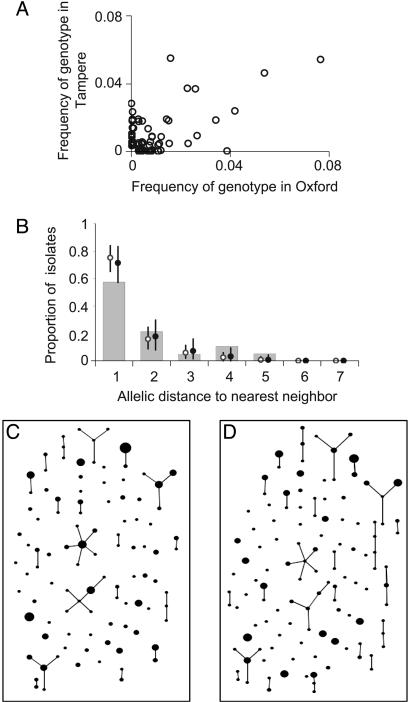
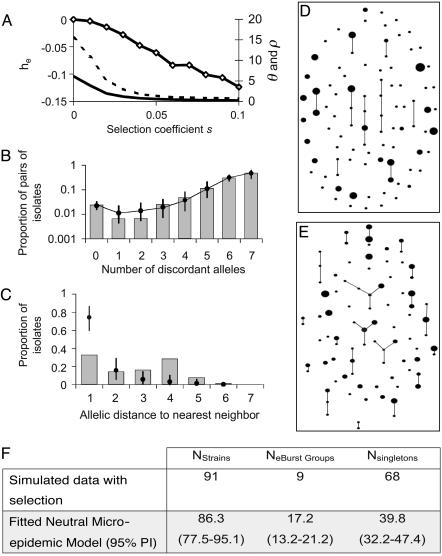
Understanding bacterial population genetics is vital for interpreting the response of bacterial populations to selection pressures such as antibiotic treatment or vaccines targeted at only a subset of strains. The evolution of transmissible bacteria occurs by mutation and localized recombination and is influenced by epidemiological as well as molecular processes. We demonstrate that the observed population genetic structure of three important human pathogens, Streptococcus pneumoniae, Neisseria meningitidis, and Staphylococcus aureus, can be explained by using a simple evolutionary model that is based on neutral mutational drift, modulated by recombination, and which incorporates the impact of epidemic transmission in local populations. The predictions of this neutral "microepidemic" model are found to closely fit observed genetic relatedness distributions of bacteria sampled from their natural population, and it provides estimates of the relative rate of recombination that agree well with empirical estimates. The analysis suggests the emergence of neutral bacterial population structure from overlapping microepidemics within clustered host populations and provides insight into the nature and size distribution of these clusters. These findings challenge the assumption that strains of bacterial pathogens differ markedly in relative fitness.
Figures
References
-
- Spratt, B. G., Hanage, W. P. & Feil, E. J. (2001) Curr. Opin. Microbiol. 4, 602–606. - PubMed
-
- Ochman, H., Lawrence, J. G. & Groisman, E. A. (2000) Nature 405, 299–304. - PubMed
-
- Supply, P., Warren, R. M., Banuls, A. L., Lesjean, S., Van Der Spuy, G. D., Lewis, L. A., Tibayrenc, M., Van Helden, P. D. & Locht, C. (2003) Mol. Microbiol. 47, 529–538. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
