

Extraction of transcription regulatory signals from genome-wide DNA-protein interaction data
- PMID: 15684410
- PMCID: PMC548334
- DOI: 10.1093/nar/gki166
Extraction of transcription regulatory signals from genome-wide DNA-protein interaction data
Abstract
Deciphering gene regulatory network architecture amounts to the identification of the regulators, conditions in which they act, genes they regulate, cis-acting motifs they bind, expression profiles they dictate and more complex relationships between alternative regulatory partnerships and alternative regulatory motifs that give rise to sub-modalities of expression profiles. The 'location data' in yeast is a comprehensive resource that provides transcription factor-DNA interaction information in vivo. Here, we provide two contributions: first, we developed means to assess the extent of noise in the location data, and consequently for extracting signals from it. Second, we couple signal extraction with better characterization of the genetic network architecture. We apply two methods for the detection of combinatorial associations between transcription factors (TFs), the integration of which provides a global map of combinatorial regulatory interactions. We discover the capacity of regulatory motifs and TF partnerships to dictate fine-tuned expression patterns of subsets of genes, which are clearly distinct from those displayed by most genes assigned to the same TF. Our findings provide carefully prioritized, high-quality assignments between regulators and regulated genes and as such should prove useful for experimental and computational biologists alike.
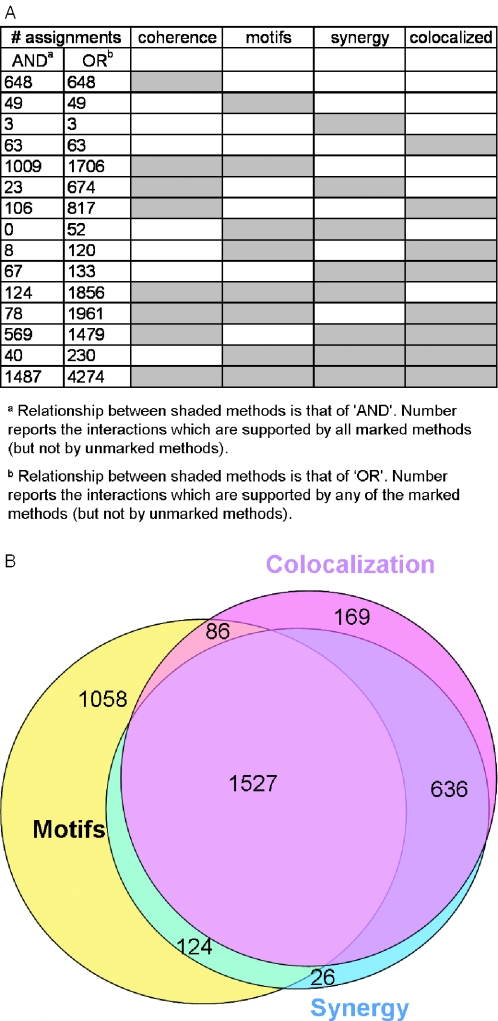
Figures
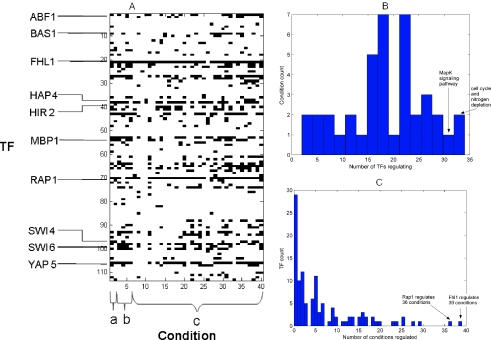
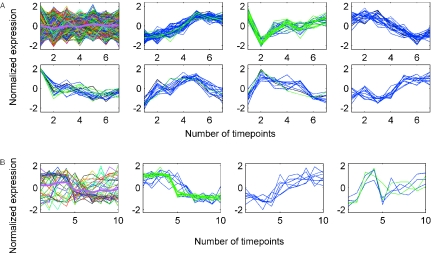
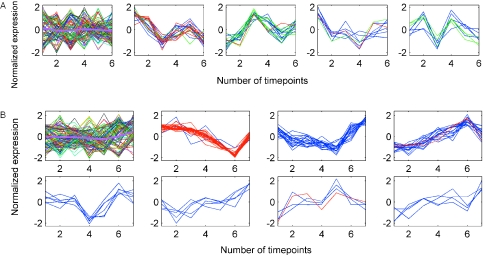
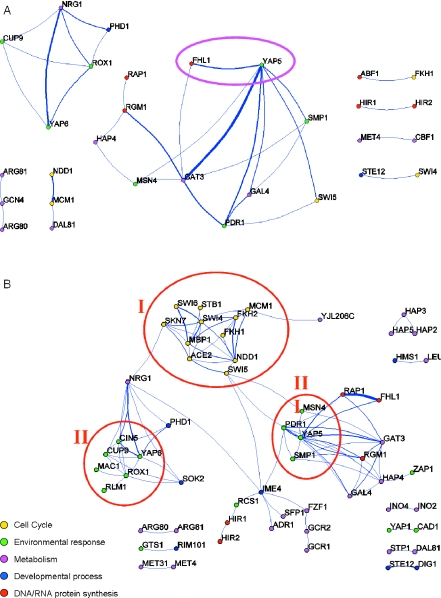
References
-
- Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K., Hannett N.M., Harbison C.T., Thompson C.M., Simon I., et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. - PubMed
-
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995;57:289–300.
-
- Segal E., Yelensky R., Koller D. Genome-wide discovery of transcriptional modules from DNA sequence and gene expression. Bioinformatics. 2003;19:i273–i282. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
